
Биоинформа́тика — междисциплинарная область, объединяющая общую биологию, молекулярную биологию, кибернетику, генетику, химию, компьютерные науки, математику и статистику. Крупномасштабные биологические проблемы, требующие анализа больших объёмов данных, решаются биоинформатикой с вычислительной точки зрения. Биоинформатика главным образом включает в себя изучение и разработку компьютерных методов и направлена на получение, анализ, хранение, организацию и визуализацию биологических данных.
Арифметическое кодирование — один из алгоритмов энтропийного сжатия.

Филогенетическое дерево — дерево, отражающее эволюционные взаимосвязи между различными видами или другими сущностями, имеющими общего предка.
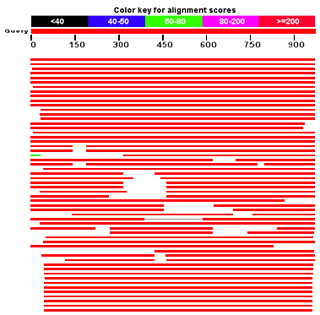
BLAST — семейство компьютерных программ, служащих для поиска гомологов белков или нуклеиновых кислот, для которых известна первичная структура (последовательность) или её фрагмент. Используя BLAST, исследователь может сравнить имеющуюся у него последовательность с последовательностями из базы данных и найти последовательности предполагаемых гомологов. Является важнейшим инструментом для молекулярных биологов, биоинформатиков и систематиков. Программа BLAST была разработана группой учёных: Стивен Альтшуль, Уоррен Гиш, Вебб Миллер, Юджин Майерс и Дэвид Липман в системе Национальных институтов здравоохранения США. Первая публикация с описанием программы вышла в Журнале молекулярной биологии в 1990 году.

Мно́жественное выра́внивание после́довательностей — выравнивание трёх и более биологических последовательностей, обычно белков, ДНК или РНК. В большинстве случаев предполагается, что входной набор последовательностей имеет эволюционную связь. Используя множественное выравнивание, можно оценить эволюционное происхождение последовательностей, проведя филогенетический анализ.
Выра́внивание после́довательностей — биоинформатический метод, основанный на размещении двух или более последовательностей мономеров ДНК, РНК или белков друг под другом таким образом, чтобы легко увидеть сходные участки в этих последовательностях. Сходство первичных структур двух молекул может отражать их функциональные, структурные или эволюционные взаимосвязи. Выровненные последовательности оснований нуклеотидов или аминокислот обычно представляются в виде строк матрицы. Добавляются разрывы между основаниями таким образом, чтобы одинаковые или похожие элементы были расположены в следующих друг за другом столбцах матрицы.
Алгоритм Нидлмана — Вунша — это алгоритм для выполнения выравнивания двух последовательностей, который используется в биоинформатике при построении выравниваний аминокислотных или нуклеотидных последовательностей. Алгоритм был предложен в 1970 году Солом Нидлманом и Кристианом Вуншем.
Предсказа́ние структу́ры белка́ — направление молекулярного моделирования, предсказание по аминокислотной последовательности трёхмерной структуры белка. Данная задача является одной из самых важных целей биоинформатики и теоретической химии. Данные, полученные при помощи предсказания, применяются в медицине и биотехнологии при создании новых ферментов).
Вычисли́тельная гено́мика использует вычислительный анализ, чтобы расшифровать последовательности генома и связанные с ними данные, включая последовательности ДНК и РНК. Также вычислительная геномика может быть определена как раздел биоинформатики, но с тем отличием, что внимание уделяется анализу полных геномов, чтобы понять принципы того, как различные ДНК управляют организмом на молекулярном уровне.

UGENE — свободное биоинформационное программное обеспечение.

Clustal — одна из самых широко используемых компьютерных программ для множественного выравнивания нуклеотидных и аминокислотных последовательностей.
Картирование коротких прочтений — биоинформатический метод анализа результатов секвенирования нового поколения, состоящий в определении позиций в референсном геноме или транскриптоме, откуда с наибольшей вероятностью могло быть получено каждое конкретное короткое прочтение. Обычно является первой стадией в обработке данных в случае, если известен геном исследуемого организма.
Количественный анализ альтернативного сплайсинга — набор экспериментальных и вычислительных методов, позволяющих определить относительные представленности различных транскриптов одного гена в биологическом образце.

Простра́нственное выра́внивание — способ установления гомологии между двумя или более полимерными структурами на основании их трёхмерной структуры. Этот процесс обычно применяется к третичной структуре белков, но может также использоваться и для больших молекул РНК. В противоположность простому наложению структур, когда известно по крайней мере несколько эквивалентных аминокислотных остатков, пространственное выравнивание не требует никаких предварительных данных, кроме координат атомов.
Моти́в в молекулярной биологии — относительно короткая последовательность нуклеотидов или аминокислот, слабо меняющаяся в процессе эволюции и, по крайней мере предположительно, имеющая определённую биологическую функцию. Под мотивом иногда подразумевают не конкретную последовательность, а каким-либо образом описанный спектр последовательностей, каждая из которых способна выполнять определённую биологическую функцию данного мотива.
Предсказа́ние втори́чной структу́ры РНК — метод определения вторичной структуры нуклеиновой кислоты по последовательности её нуклеотидов. Вторичную структуру можно предсказывать для единичной последовательности или анализировать множественное выравнивание семейства родственных РНК.

Логотип последовательностей — метод графического представления консервативности нуклеотидов или аминокислот. Логотип строится по набору выровненных последовательностей. Этот метод позволяет на одном графике отразить следующие характеристики анализируемого участка:
- консенсусную последовательность выравнивания;
- относительные частоты встречаемости элементов в каждой позиции последовательности;
- информационное содержание каждой позиции в последовательности ;
- наличие специфического локуса.
BWA — программный пакет для картирования коротких прочтений на большие референсные геномы, написанный китайским биоинформатиком Хенг Ли и англичанином Ричардом Дурбиным. Является одним из широкоиспользуемых алгоритмов выравнивания, а также рекомендуется для анализа данных производителями Illumina. BWA состоит из трёх основных алгоритмов: BWA-BackTrack, BWA-SW и BWA-MEM. В основе алгоритмов BWA лежит преобразование Барроуза—Уилера, суффиксные массивы и алгоритм выравнивания Смита—Ватермана. Программный пакет умеет работать с длинными последовательностями на порядок быстрее, чем MAQ при достижении аналогичной точности выравнивания.
Множество Смита — Вольтерры — Кантора — пример множества точек на вещественной оси  , которое нигде не плотно, но, однако, имеет положительную меру. Топологически эквивалентно классическому канторову множеству. Названо по именам математиков Генри Смита, Вито Вольтерры и Георга Кантора.
, которое нигде не плотно, но, однако, имеет положительную меру. Топологически эквивалентно классическому канторову множеству. Названо по именам математиков Генри Смита, Вито Вольтерры и Георга Кантора.
Завистливое разрезание торта — вид справедливого разрезания торта. Это разрезание неоднородного ресурса («торта») с удовлетворением критерия отсутствия зависти, а именно, что любой участник обладает чувством, что выделенная ему часть не меньше кусков, отданных другим участникам.