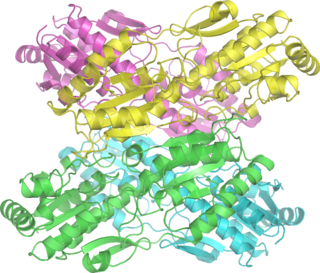
Альдолаза B
| Альдолаза B | |||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
 | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Идентификаторы | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Псевдонимы | ALDOBaldolase Bfructose-bisphosphatasealdolase 2fructose-bisphosphate aldolase Bliver-type aldolasealdolase Bfructose-bisphosphate | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Внешние ID | GeneCards: [1] | ||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||

Альдолаза B, также фруктозо-бисфосфат альдолаза B или реже печёночный тип альдолазы (англ. Aldolase B, сокр. ALDOB) — фермент (КФ 4.1.2.13) из семейства фруктозо-1,6-бисфосфат альдолазы I класса (класс лиазы), является одним из трёх изоферментов (A, B и C). Альдолаза B играет ключевую роль в углеводном обмене: в процессах гликолиза и глюконеогенеза, а также в метаболизме фруктозы. Фермент катализирует реакцию альдольного (негидролитического) расщепления связей C-C в молекуле фруктозо-1,6-бисфосфата (Ф-1,6-БФ) до дигидроксиацетонфосфата (ДАФ) и глицеральдегид-3-фосфата (ГАФ), обратную реакцию — альдольную конденсацию, а также расщепление фруктозо-1-фосфата на глицеральдегид и дигидроксиацетонфосфат, и их альдольную конденсацию во фруктозо-1-фосфат. Схема реакции, катализируемой альдолазой B:
- фруктозо-1,6-бисфосфат
 дигидроксиацетонфосфат + глицеральдегид-3-фосфат
дигидроксиацетонфосфат + глицеральдегид-3-фосфат - фруктозо-1-фосфат дигидроксиацетонфосфат + глицеральдегид.
У млекопитающих (включая и человека) альдолаза B преимущественно экспрессируется в печени. Субстратная специфичность альдолазы В такова, что она примерно в одинаковом соотношении способна катализировать расщепление и конденсацию молекул, как фруктозо-1,6-бисфосфата, так и фруктозо-1-фосфата (до глицеральдегида и дигидроксиацетонфосфата), в то же время альдолазы A и C, предпочитают первый субстрат (фруктозо-1,6-бисфосфат)[1]. Различия в субстратной специфичности альдолазы B обусловлены структурными особенностями данного фермента, она закодирована другим геном. У человека ген, кодирующий альдолазу B — ALDOB, локализован на длинном плече (q-плече) 9-й хромосомы. Ген имеет длину 14 500 пар оснований и содержит 9 экзонов[2][3][4]. Дефекты этого гена были идентифицированы как причина наследственной непереносимости фруктозы.
Структура
Альдолаза B представляет собой гомотетрамерный фермент, состоящий из четырёх субъединиц с молекулярной массой 36 кДа и локальной симметрией 222. Каждая субъединица имеет молекулярную массу 36 кДа и содержит восьмицепочечный α/β-цилиндр, который окружает лизин-229 (данная аминокислота, образует основание Шиффа, которое является ключевой для катализа)[5][6].
Изофермент-специфические области
Несмотря на то, что большая часть общей структуры фермента альдолазы является консервативной для всех трёх изоферментов, было установлено, что четыре области общего фермента альдолазы сильно различаются между изоферментами. Такие области были названы изофермент-специфичными (ISR1-4). Считается, что именно эти участки придают изоферментам специфичность и структурные различия. Все ISR 1-3 находятся в экзоне 3 гена ALDOB. ISR 4 является наиболее вариабельным из четырёх и находится на с-концевой части белка[1].
ISR 1-3 обнаруживаются преимущественно в участках на поверхности фермента. Эти участки не перекрываются с активным сайтом, что указывает на то, что ISRs могут изменять субстратную специфичность изофермента на расстоянии или вызывать взаимодействие С-конца с активным сайтом[6]. Согласно недавно выдвинутой теории, ISRs могут обеспечивать различную конформационную динамику фермента альдолазы, которая объясняет её специфичность[7].
Механизм катализа
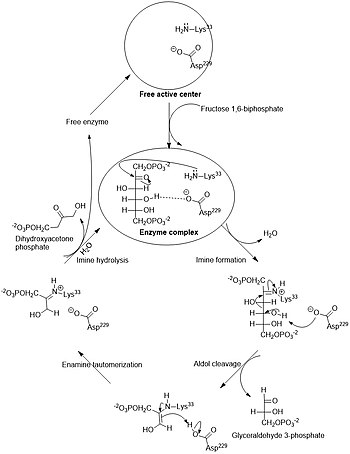
Общий фермент фруктозо-бисфосфат альдолаза расщепляет 6-углеродный сахар фруктозу на два 3-углеродных продукта в реакции ретроальдольной конденсации (обратная альдольной конденсации). Для этой реакции характерно образование интермедиата (промежуточного соединения) основания Шиффа с остатком лизина (лизин-229) в активном сайте фермента; образование основания Шиффа является ключевым отличием альдолаз I типа (продуцируемых животными) от альдолаз II типа (продуцируемых грибами и бактериями). После образования основания Шиффа четвёртая гидроксильная группа на фруктозном остове депротонируется остатком аспартата (аспартат-33), что приводит к альдольному расщеплению. При гидролизе основания Шиффа образуются два трёхуглеродных продукта. В зависимости от реактанта, Ф-1-Ф или Ф-1,6-БФ, продуктами являются ДАФ и глицеральдегид или глицеральдегид-3-фосфат, соответственно[8].
ΔG°' этой реакции составляет +23,9 кДж/моль (реакция — эндергоническая). Хотя эта реакция может показаться слишком сложной для протекания, следует отметить, что в физиологических условиях ΔG этой реакции падает до значения, близкого к нулю или ниже. Например, ΔG этой реакции в физиологических условиях в эритроцитах составляет -0,23 кДж/моль[8].
Физиология
Альдолаза В играет ключевую роль в углеводном обмене, поскольку катализирует один из основных этапов гликолитико-глюконеогенного пути. Хотя она катализирует расщепление глюкозы, особенно важную роль она играет в метаболизме фруктозы, который происходит в основном в печени, в корковом слое почек и слизистой оболочке тонкого кишечника. При всасывании фруктозы она фосфорилируется фруктокиназой с образованием фруктозо-1-фосфата (Ф-1-Ф). Затем альдолаза В катализирует распад Ф-1-Ф на глицеральдегид и дигидроксиацетонфосфат. После того как глицеральдегид фосфорилируется триозокиназой с образованием глицеральдегид-3-фосфата, оба продукта могут быть использованы в гликолитико-глюконеогенном пути, то есть модифицированы в глюкозу или пируват[9].
Хотя механизм регуляции альдолазы В неизвестен, было отмечено усиление транскрипции гена ALDOB в печени животных при увеличении количества углеводов в рационе и снижении концентрации глюкагона[10][11].
Патология
Мутации в гене ALDOB, приводящие к дефектам альдолазы В, вызывают заболевание, называемое наследственной непереносимостью фруктозы. Из-за отсутствия функциональной альдолазы В организмы с наследственной непереносимостью фруктозы не могут должным образом утилизировать фруктозо-1-фосфат (Ф-1-Ф), что приводит к накоплению Ф-1-Ф в тканях организма. Высокий уровень Ф-1-Ф не только токсичен для клеток тканей, но и удерживает фосфат в непригодной для использования форме, которая не возвращается в общий фосфатный пул, что приводит к истощению запасов фосфатов и АТФ. Отсутствие легкодоступного фосфата приводит к прекращению гликогенолиза в печени, что приводит к гипогликемии[12]. Такое накопление Ф-1-Ф также ингибирует глюконеогенез, что ещё больше снижает количество легкодоступной глюкозы. Потеря АТФ приводит к множеству проблем, включая подавление синтеза белка, печёночную и почечную дисфункцию. Однако в случаях наследственной непереносимости фруктозы прогноз пациентов благоприятный. Отказ от продуктов, содержащих фруктозу, сахарозу и сорбит, позволяет пациентам жить без симптомов[9].
Наследственная непереносимость фруктозы (ННФ) — врождённое заболевание с аутосомно-рецессивным типом наследованием. Идентифицировано около 30 мутаций, вызывающих ННФ, и их совокупность приводит к тому, что частота заболевания составляет 1 на каждые 20 000 рождений[9][13]. Мутантные аллели являются результатом различных типов мутаций, включая замены пар оснований и небольшие делеции. Наиболее распространённой мутацией является A149P, представляющая собой трансверсию гуанина в цитозин в экзоне 5, в результате чего аланин в положении 149 заменяется на пролин. На долю этого специфического мутантного аллеля приходится 53 % аллелей ННФ[14]. Другие мутации, приводящие к ННФ, встречаются реже и часто коррелируют с наследственным происхождением[15].
Примечания
- ↑ 1 2 Dalby AR, Tolan DR, Littlechild JA (November 2001). "The structure of human liver fructose-1,6-bisphosphate aldolase". Acta Crystallogr. D. 57 (Pt 11): 1526—33. doi:10.1107/S0907444901012719. PMID 11679716.
- ↑ Entrez Gene: ALDOB aldolase B, fructose-bisphosphate.
- ↑ Henry I, Gallano P, Besmond C, Weil D, Mattei MG, Turleau C, Boué J, Kahn A, Junien C (July 1985). "The structural gene for aldolase B (ALDB) maps to 9q13----32". Ann. Hum. Genet. 49 (Pt 3): 173—80. doi:10.1111/j.1469-1809.1985.tb01691.x. PMID 3000275. S2CID 10058239.
- ↑ Tolan DR, Penhoet EE (June 1986). "Characterization of the human aldolase B gene". Mol. Biol. Med. 3 (3): 245—64. PMID 3016456.
- ↑ Sygusch J, Beaudry D, Allaire M (November 1987). "Molecular architecture of rabbit skeletal muscle aldolase at 2.7-A resolution". Proc. Natl. Acad. Sci. U.S.A. 84 (22): 7846—50. doi:10.1073/pnas.84.22.7846. PMC 299418. PMID 3479768.
- ↑ 1 2 Pezza JA, Choi KH, Berardini TZ, Beernink PT, Allen KN, Tolan DR (May 2003). "Spatial clustering of isozyme-specific residues reveals unlikely determinants of isozyme specificity in fructose-1,6-bisphosphate aldolase". J. Biol. Chem. 278 (19): 17307—13. doi:10.1074/jbc.M209185200. PMID 12611890.
- ↑ Pezza JA, Stopa JD, Brunyak EM, Allen KN, Tolan DR (November 2007). "Thermodynamic Analysis Shows Conformational Coupling/Dynamics Confers Substrate Specificity in Fructose-1,6-bisphosphate Aldolase". Biochemistry. 46 (45): 13010—8. doi:10.1021/bi700713s. PMC 2546497. PMID 17935305.
- ↑ 1 2 Biochemistry. — 4th. — Brooks/Cole, 2010.
- ↑ 1 2 3 Inborn Metabolic Diseases. — Fourth Revised. — Springer Berlin Heidelberg, 2006.
- ↑ Gomez PF, Ito K, Huang Y, Otsu K, Kuzumaki T, Ishikawa K (November 1994). "Dietary and hormonal regulation of aldolase B gene transcription in rat liver". Arch Biochem Biophys. 314 (2): 307—14. doi:10.1006/abbi.1994.1447. PMID 7979370.
- ↑ Munnich A, Besmond C, Darquy S, et al. (March 1985). "Dietary and hormonal regulation of aldolase B gene expression". J. Clin. Invest. 75 (3): 1045—52. doi:10.1172/JCI111766. PMC 423659. PMID 2984252.
- ↑ Bouteldja N, Timson DJ (April 2010). "The biochemical basis of hereditary fructose intolerance". J. Inherit. Metab. Dis. 33 (2): 105—12. doi:10.1007/s10545-010-9053-2. PMID 20162364. S2CID 207099820.
- ↑ Esposito G, Vitagliano L, Santamaria R, Viola A, Zagari A, Salvatore F (November 2002). "Structural and functional analysis of aldolase B mutants related to hereditary fructose intolerance". FEBS Lett. 531 (2): 152—6. doi:10.1016/S0014-5793(02)03451-8. PMID 12417303. S2CID 7134716.
- ↑ Malay AD, Allen KN, Tolan DR (March 2005). "Structure of the thermolabile mutant aldolase B, A149P: molecular basis of hereditary fructose intolerance". J Mol Biol. 347 (1): 135—44. doi:10.1016/j.jmb.2005.01.008. PMID 15733923.
- ↑ Tolan DR (1995). "Molecular basis of hereditary fructose intolerance: mutations and polymorphisms in the human aldolase B gene". Hum. Mutat. 6 (3): 210—8. doi:10.1002/humu.1380060303. PMID 8535439. S2CID 35127545.
Внешние ссылки
| Гликолиз | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Только глюконеогенез |
| ||||||||
| Регуляция | |||||||||