
Боле́знь Паркинсо́на — медленно прогрессирующее хроническое нейродегенеративное неврологическое заболевание, характерное для лиц старшей возрастной группы. Относится к дегенеративным заболеваниям экстрапирамидной моторной системы. Вызвано прогрессирующим разрушением и гибелью нейронов, вырабатывающих нейромедиатор дофамин, — прежде всего в чёрной субстанции, а также и в других отделах центральной нервной системы. Недостаточная выработка дофамина ведёт к тормозному влиянию базальных ганглиев на кору головного мозга. Ведущими симптомами являются:
- мышечная ригидность;
- гипокинезия;
- тремор;
- постуральная неустойчивость.

Тригонометри́ческие фу́нкции — элементарные функции, которые исторически возникли при рассмотрении прямоугольных треугольников и выражали зависимости длин сторон этих треугольников от острых углов при гипотенузе. Эти функции нашли широкое применение в самых разных областях науки. По мере развития математики определение тригонометрических функций было расширено, в современном понимании их аргументом может быть произвольное вещественное или комплексное число.
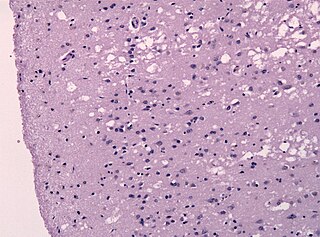
Прио́ны — особый класс инфекционных патогенов, не содержащих нуклеиновых кислот. Прионы представляют собой белки с аномальной третичной структурой. Это положение лежит в основе прионной гипотезы, однако насчёт состава прионов существуют и другие точки зрения.
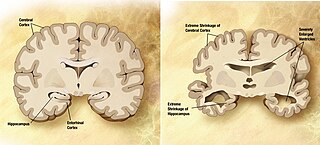
Боле́знь Альцгеймера — наиболее распространённая форма деменции, нейродегенеративное заболевание, впервые описанное в 1907 году немецким психиатром Алоисом Альцгеймером (1864—1915). Как правило, она обнаруживается у людей старше 65 лет, но существует и ранняя болезнь Альцгеймера — редкая форма заболевания. Общемировая заболеваемость на 2006 год оценивалась в 26,6 млн человек, а к 2050 году число больных может вырасти вчетверо.

Болезнь Гентингтона — аутосомно-доминантное генетическое заболевание нервной системы, характеризующееся постепенным началом обычно в возрасте 30—50 лет и сочетанием прогрессирующего хореического гиперкинеза и психических расстройств. Заболевание вызывается умножением кодона CAG в гене HTT. Этот ген кодирует 350-kDa белок хантингтин с неизвестной функцией. В гене дикого типа у разных людей присутствует разное количество CAG-повторов, однако, когда число повторов превышает 36, развивается болезнь. Нейроморфологическая картина характеризуется атрофией стриатумa, а на поздней стадии также атрофией коры головного мозга.

Гликозил-гидролазы катализируют гидролиз гликозидных связей в молекулах углеводов, приводя к появлению двух более мелких молекул углеводов. Эти ферменты встречаются в клетках почти всех живых организмов. Гликозидазы выполняют множество разнообразных функций: деградация биомассы, участие в антибактериальной защите организма, развитие патогенеза, клеточный биосинтез. Гликозидазы вместе с гликозилтрансферазами образуют основу биологического аппарата синтеза и разрушения гликозидных связей.

Альфа-галактозида́за, мелибиаза (melibiase) — фермент группы гликозил-гидролаз, способный отщеплять терминальные нередуцирующие остатки α-D-галактозы от α-D-галактозидов, как правило, с сохранением их оптической конфигурации, в частности, от галактозных олигосахаридов, галактоманнанов и галактолипидов.

Боле́знь Тея — Са́кса (GM2 ганглиозидоз, ранняя детская амавротическая идиотия) — редкое наследственное заболевание с аутосомно-рецессивным типом наследования, поражающее центральную нервную систему (спинной и головной мозг, а также менингеальные оболочки). Относится к группе лизосомных болезней накопления. Названо в честь британского офтальмолога Уоррена Тея (англ. Warren Tay, 1843—1928) и американского невролога Бернарда Сакса (англ. Bernard Sachs, 1858—1944), которые впервые описали это заболевание независимо друг от друга в 1881 и 1887 годах соответственно.
Фа́ктор некро́за о́пухоли — внеклеточный белок, многофункциональный провоспалительный цитокин, синтезирующийся в основном моноцитами и макрофагами. Влияет на липидный метаболизм, коагуляцию, устойчивость к инсулину, функционирование эндотелия, стимулирует продукцию ИЛ-1, ИЛ-6, ИЛ-8, интерферона-гамма, активирует лейкоциты, один из важных факторов защиты от внутриклеточных паразитов и вирусов. Впервые был обнаружен в сыворотке мышей, которым были введены БЦЖ и эндотоксин. Сыворотка из таких мышей обладала цитотоксическим и цитостатическим действием на некоторые трансформированные клеточные линии, а также вызывала геморрагический некроз и уменьшение привитых опухолей у мышей. Активирует ядерный транскрипционный фактор NF-κB. Избыточная продукция ФНО вызывает расстройства гемодинамики, цитотоксический эффект на клетки организма. Нарушения регуляции ФНО у человека ассоциированы с различными заболеваниями, такими как болезнь Альцгеймера, рак, клиническая депрессия, псориаз и воспалительные заболевания кишечника.
3-я хромосо́ма челове́ка — одна из 24 человеческих хромосом, одна из 22 аутосом человека. Хромосома содержит почти 200 млн пар оснований, что составляет примерно 6,5 % всего материала ДНК человеческой клетки. В настоящее время считается, что на 3-й хромосоме находятся 1960 генов.
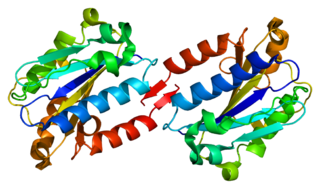
Интегрин альфа-L — мембранный белок, гликопротеин из надсемейства интегринов, продукт гена ITGAL (CD11A), альфа-субъединица интегрина αLβ2 (LFA-1).
Нейродегенеративные заболевания — группа в основном медленно прогрессирующих, наследственных или приобретённых заболеваний нервной системы. Общим для этих заболеваний является прогрессирующая гибель нервных клеток (нейродегенерация), ведущая к различным неврологическим симптомам — прежде всего, к деменции и нарушению движений. Заболевания могут наступить в различном возрасте, протекают диффузно или генерализированно, гистологически определяется специфический тип изменений.

Лизосо́мные боле́зни накопле́ния — общее название группы весьма редких наследственных заболеваний, вызванных нарушением функции внутриклеточных органелл лизосом. Эти одномембранные органоиды являются частью эндомембранной системы клетки и специализируются на внутриклеточном расщеплении веществ: гликогена, гликозаминогликанов, гликопротеинов и других. Лизосомные болезни накопления вызываются генетически обусловленным дефицитом ферментов лизосом, что приводит к накоплению макромолекул, являющихся субстратом этих ферментов, в различных органах и тканях организма.

Боле́знь По́мпе — редкое, наследственное заболевание с аутосомно-рецессивным механизмом, наследования,, связанное с повреждением мышечных, и нервных клеток по всему организму. Клиническая картина данной патологии, обусловлена накоплением гликогена, в лизосомах, вызванным недостаточностью лизосомного фермента — кислой α-1,4-глюкозидазы. Различают быстро прогрессирующую (классическую) и медленно прогрессирующую формы болезни Помпе. Несмотря на широкий спектр клинических проявлений, гликогеноза II типа, в основе всех форм болезни лежит, дефицит одного фермента, кодируемого геном GAA.

Боле́знь Фабри́ — редкое генетически детерминированное заболевание с Х-сцепленным типом наследования, из группы лизосомных болезней накопления. Ранее считалось, что тип наследования болезни Фабри — X-сцепленный рецессивный, однако на современном этапе накоплено достаточно данных, чтобы считать тип наследования болезни Фабри X-сцепленным доминантным с неполной пенетрантностью у женщин. Данное заболевание вызвано нарушением метаболизма сфинголипидов и обладает широким спектром клинических симптомов.

Синдром Сандхоффа — наследственное заболевание, вариант 0 GM2-ганглиозидоза из группы лизосомных болезней накопления — нарушение липидного обмена, вызванное наследственным дефицитом (недостаточностью) ферментов бета-гексозаминидазы А и Б.

Бета-маннозидоз — редкое аутосомно-рецессивное наследственное заболевание из группы лизосомных болезней накопления, связанное с нарушением метаболизма олигосахаридов в результате снижения активности фермента лизосом бета-маннозидазы.

Фукозидо́з — редкое наследственное заболевание из группы лизосомных болезней накопления, связанное с недостаточностью гидролаз, расщепляющих полисахаридные связи. В результате внутри клеток накапливаются два класса макромолекул: гликолипиды и гликопротеины. Клиническая картина связана с мутацией, приводящей к повреждению или дефициту фермента лизосом альфа-L-фукозидазы и характеризуется разнообразными соматическими проявлениями и нарушением функции нервной системы.
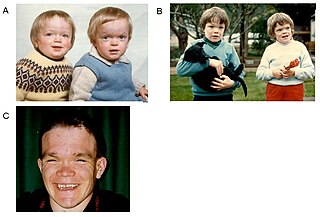
Альфа-маннозидо́з — редкое аутосомно-рецессивное наследственное заболевание из группы лизосомных болезней накопления, связанное с нарушением метаболизма олигосахаридов в результате снижения активности фермента лизосом альфа-маннозидазы. Также заболевание наносит ущерб в животноводстве.

Убиквитин карбокси-концевая гидролаза L1 представляет собой деубиквитинирующий фермент.