Апоптоз

Микрофотография, сделанная с помощью
сканирующего электронного микроскопа

Апопто́з (др.-греч. ἀπόπτωσις «опадание», от ἀπό- + πτῶσις «падение») — регулируемый процесс программируемой клеточной гибели, в результате которого клетка распадается на отдельные апоптотические тельца, ограниченные плазматической мембраной. Фрагменты погибшей клетки обычно очень быстро (в среднем за 90 минут[1]) фагоцитируются макрофагами либо соседними клетками, минуя развитие воспалительной реакции. Морфологически регистрируемый процесс апоптоза продолжается 1—3 часа[2]. Одной из основных функций апоптоза является уничтожение дефектных (повреждённых, мутантных, инфицированных) клеток. В многоклеточных организмах апоптоз к тому же задействован в процессах дифференциации и морфогенеза, в поддержании клеточного гомеостаза, в обеспечении важных аспектов развития и функционирования иммунной системы. Апоптоз наблюдается у всех эукариот, начиная от одноклеточных простейших и вплоть до высших организмов. В программируемой смерти прокариот участвуют функциональные аналоги эукариотических белков апоптоза[3].
Исследования программируемой клеточной смерти ведутся с конца 1960-х годов. Термин «апоптоз» был впервые употреблён в 1972 году в работе британских учёных — Дж. Керра, Э. Уайли и А. Керри. Одними из первых к изучению генетики и молекулярных механизмов апоптоза приступили С. Бреннер, Дж. Салстон и Р. Хорвиц, все трое в 2002 году были удостоены Нобелевской премии по физиологии или медицине за открытия в области генетической регуляции развития органов и за достижения в исследованиях программируемой клеточной смерти. В настоящее время установлены основные механизмы реализации апоптоза в эукариотических клетках, активно ведутся исследования регуляторов и активаторов апоптоза. Интерес учёных связан с возможностью применения знаний о программируемой клеточной смерти в медицине при лечении онкологических, аутоиммунных и нейродегенеративных заболеваний.[4]
В организме среднестатистического взрослого человека в результате апоптоза погибает ежедневно порядка 50—70 миллиардов клеток. Для среднестатистического ребёнка в возрасте от 8 до 14 лет число клеток, погибших путём апоптоза, составляет порядка 20—30 миллиардов в день. Суммарная масса клеток, которые на протяжении 1 года жизни подвергаются разрушению, эквивалентна массе тела человека. При этом восполнение утраченных клеток обеспечивается за счёт пролиферации — увеличения клеточной популяции путём деления.[5]
История исследования

Прогрессивное развитие методов изучения клетки к концу XIX века было сопряжено с важными открытиями в области цитологии. Тем не менее регистрируемые факты клеточной смерти в рамках представлений той эпохи считались случайными и незапланированными явлениями.[7] К примеру, упоминание факта клеточной смерти встречается в работе Карла Фохта, датированной 1842 годом.[8][9]
Более пристальное внимание процессу клеточной гибели было уделено во второй половине XX столетия. В 1951 году была опубликована статья Альфреда Глюксманна, в которой клеточная смерть была рассмотрена в контексте нормального онтогенетического развития.[7][8][9] Исследования феномена программируемой клеточной смерти начались с конца 1960-х годов. Одним из первых в данной области был Джон Керр, который изучал гибель гепатоцитов при остром поражении печени у крыс.[4] В 1972 году коллектив британских учёных во главе с Дж. Керром впервые предложили использовать термин «апоптоз» для обозначения программируемой клеточной смерти. В своей статье, опубликованной в «Британском журнале онкологических исследований[англ.]», они дали морфологическое описание апоптоза и высказали основные представления о функциях данного процесса.[10] 14 марта 2000 года Джон Керр был удостоен престижной премии Пауля Эрлиха и Людвига Дармштедтера за свой вклад в исследования апоптоза.[4]
В 1974 году трое учёных из кембриджской лаборатории молекулярной биологии — Сидней Бреннер, Джон Салстон и Роберт Хорвиц — приступили к изучению развития клеток нематоды Caenorhabditis elegans. В ходе их исследований выяснилось, что при развитии C. elegans погибает 131 из 1090 клеток. Со временем учёным удалось выявить генетические детерминанты и молекулярные механизмы апоптоза.[11] В итоге С. Бреннер, Дж. Салстон и Р. Хорвиц в 2002 году были удостоены Нобелевской премии по физиологии или медицине за открытия в области генетической регуляции развития органов и за достижения в исследованиях программируемой клеточной смерти.[12] Помимо этого в 2000 году Роберту Хорвицу наряду с Джоном Керром была вручена премия Пауля Эрлиха и Людвига Дармштедтера.[4]
Начиная с последней декады XX века наблюдается интенсивное увеличение числа публикаций, посвящённых исследованиям апоптоза. В 1988 году было установлено, что Bcl-2 ингибирует процесс апоптоза.[13] С конца 1980-х годов начались первые работы по исследованию Fas/FasL-рецепторно/лигандной системы, лежащей в основе одного из основных рецептор-зависимых путей апоптоза.[14] В 1992 году установлен факт экспрессии фосфатидилсерина на внешней мембране апоптотических клеток. В 1993 году были идентифицированы ингибиторы белков апоптоза (IAP). В 1994 году начаты исследования структуры каспаз.[13] В 1996 году было установлено, что цитохром c в комплексе с АТФ участвует в активации каспазы-3.[15]
К 2000 году число публикаций на тему апоптоза превышало 35 539[4], а уже к марту 2008 года количество публикаций превысило отметку в 143 400.[6] В настоящее время установлены основные механизмы реализации апоптоза в эукариотических клетках, активно ведутся исследования регуляторов и активаторов апоптоза. Интерес учёных связан с возможностью применения знаний о программируемой клеточной смерти при лечении онкологических, аутоиммунных и нейродегенеративных заболеваний.[4][6]
Фазы апоптоза
Процесс апоптоза можно условно разделить на три фазы: сигнальную (индукторную), эффекторную и деградационную (фаза экзекуции или деструкции)[1][17][18].
Сигнальная фаза
Инициация апоптоза может происходить посредством внешних (внеклеточных) или внутриклеточных факторов. Например, в результате гипоксии, гипероксии, субнекротического поражения химическими или физическими агентами, перекрёстного связывания соответствующих рецепторов, нарушения сигналов клеточного цикла, удаления факторов роста и метаболизма и т. д.[17] Несмотря на разнообразие инициирующих факторов, выделяются два основных пути передачи сигнала апоптоза: рецептор-зависимый (внешний) сигнальный путь с участием рецепторов гибели клетки и митохондриальный (собственный) путь[19][20].
Рецептор-зависимый сигнальный путь
Процесс апоптоза часто (например, у млекопитающих) начинается с взаимодействия специфических внеклеточных лигандов[~ 1] с рецепторами клеточной гибели, экспрессированными на поверхности клеточной мембраны. Рецепторы, воспринимающие сигнал апоптоза, относятся к суперсемейству TNF-рецепторов (англ. tumor necrosis factor receptor или кратко TNFR — «рецептор фактора некроза опухолей»)[3][19]. Наиболее изученными рецепторами смерти, для которых описана и определена роль в апоптозе, являются CD95 (также известный как Fas или APO-1) и TNFR1 (также называемый p55 или CD120a). К дополнительным относятся CARI, DR3 (англ. death receptor 3 — «рецептор смерти 3»), DR4 и DR5.
Все рецепторы смерти представляют собой трансмембранные белки, характеризующиеся наличием общей последовательности из 80 аминокислот в цитоплазматическом домене. Данная последовательность называется доменом смерти (англ. death domain или кратко DD) и является необходимой для трансдукции сигнала апоптоза[19]. Внеклеточные участки рецепторов смерти взаимодействуют с тримерами лигандов (CD95L, TNF, Apo3L, Apo2L и т. п.). Тримеры лигандов в результате взаимодействия тримеризуют рецепторы смерти (то есть «сшивают» 3 молекулы рецептора)[21]. Активированный таким образом рецептор взаимодействует с соответствующим внутриклеточным адаптером (или адаптерами). Для рецептора CD95(Fas/APO-1) адаптером является FADD (от англ. Fas-associated DD-protein — «белок, взаимодействующий с доменом смерти Fas-рецептора»). Для рецепторов TNFR1 и DR3 адаптером является TRADD (от англ. TNFR1-associated DD-protein — «белок, взаимодействующий с доменом смерти TNFR1-рецептора»).
Адаптер, ассоциированный с рецептором смерти, вступает во взаимодействие с эффекторами — пока ещё неактивными предшественниками протеаз из семейства инициирующих каспаз — с прокаспазами. В результате цепочки взаимодействия «лиганд-рецептор-адаптер-эффектор» формируются агрегаты, в которых происходит активация каспаз. Данные агрегаты именуются апоптосомами, апоптозными шаперонами или сигнальными комплексами, индуцирующими смерть (от англ. DISC — death-inducing signaling complex — «сигнальный комплекс, индуцирующий смерть»). Примером апоптосомы может служить комплекс FasL-Fas-FADD-прокаспаза-8, в котором активируется каспаза-8[22][23].
Рецепторы смерти, адаптеры и эффекторы взаимодействуют между собой сходными по структуре доменами: DD, DED, CARD. DD (от англ. death domain — «домен смерти[англ.]») участвует во взаимодействии рецептора Fas с адаптером FADD и во взаимодействии рецепторов TNFR1 или DR3 с адаптером TRADD. Посредством домена DED (от англ. death-effector domain — «домен эффектора смерти») осуществляется взаимодействие адаптера FADD с прокаспазами −8 и −10. Домен CARD (от англ. caspase activation and recruitment domain — «домен активации и рекрутирования каспазы») участвует во взаимодействии адаптера RAIDD с прокаспазой-2[22].
Посредством рецепторов смерти могут быть активированы три инициирующие каспазы: −2; −8 и −10[22]. Активированные инициирующие каспазы далее участвуют в активации эффекторных каспаз.

«Цитохром c — APAF-1 — CARD — прокаспаза-9»[24]
Митохондриальный сигнальный путь
Большинство форм апоптоза у позвоночных реализуется по митохондриальному пути, а не через рецепторы клеточной гибели[25]. Митохондриальный сигнальный путь апоптоза реализуется в результате выхода апоптогенных белков из межмембранного пространства митохондрий в цитоплазму клетки. Высвобождение апоптогенных белков, предположительно, может осуществляться двумя путями: за счёт разрыва митохондриальной мембраны или же путём открытия высокопроницаемых каналов на внешней мембране митохондрий[22][26].
Ключевым событием митохондриального пути апоптоза является повышение проницаемости наружной мембраны митохондрий (англ. mitochondrial outer membrane permeabilization, MOMP)[25]. Существенную роль в повышении MOMP играют апоптотические Bcl-2-белки — Bax и Bak. Они встраиваются в наружную мембрану митохондрий и олигомеризуются. При этом, вероятно, нарушается целостность внешней мембраны митохондрий, по неизвестному пока механизму[27]. При повышении MOMP из межмембранного пространства митохондрий в цитозоль высвобождаются растворимые белки, участвующие в апоптозе: цитохром c — белок с молекулярной массой 15 кДа; прокаспазы −2, −3 и −9; белок AIF (англ. apoptosis inducing factor — «фактор, индуцирующий апоптоз») — флавопротеин с молекулярной массой 57 кДа[22].
Разрыв внешней мембраны митохондрий объясняется увеличением объёма митохондриального матрикса. Данный процесс связывают с раскрытием пор митохондриальной мембраны, приводящим к снижению мембранного потенциала и высокоамплитудному набуханию митохондрий вследствие осмотического дисбаланса. Поры диаметром 2,6—2,9 нм способны пропускать низкомолекулярные вещества массой до 1,5 кДа.[3][22][28] Раскрытие пор стимулируют следующие факторы: неорганический фосфат; каспазы; SH-реагенты; истощение клеток восстановленным глутатионом; образование активных форм кислорода; разобщение окислительного фосфорилирования протонофорными соединениями; увеличение содержания Ca2+ в цитоплазме; воздействие церамида; истощение митохондриального пула АТФ и др.[22][28]

Цитохром c в цитоплазме клетки участвует в формировании апоптосомы вместе с белком APAF-1 (от англ. Apoptosis Protease Activating Factor-1 — «активирующий фактор апоптотической протеазы-1»). Предварительно APAF-1 претерпевает конформационные изменения в результате реакции, протекающей с затратой энергии АТФ. Предполагается, что трансформированный APAF-1 приобретает способность связывать цитохром c. К тому же открывается доступ CARD-домена APAF-1 для прокаспазы-9. В итоге происходит олигомеризация 7 субъединиц трансформированного белка APAF-1 с участием цитохрома c и прокаспазы-9.[24] Так образуется апоптосома, активирующая каспазу-9. Зрелая каспаза-9 связывает и активирует прокаспазу-3 с образованием эффекторной каспазы-3.[22] Высвобождающийся из межмембранного пространства митохондрий флавопротеин AIF является эффектором апоптоза, действующим независимо от каспаз.[22][30]
Другие пути индукции апоптоза
Реализация апоптоза может происходить в результате комбинированного действия двух основных сигнальных путей — рецептор-зависимого и митохондриального.[31] Помимо этого, существует ряд менее распространённых механизмов инициации апоптоза. Например, за счёт активации прокаспазы-12, локализованной в эндоплазматическом ретикулуме. Высвобождение и активация прокаспазы-12 при этом обусловлены нарушениями внутриклеточного гомеостаза ионов кальция (Ca2+).[32] Активация апоптоза также может быть связана с нарушением адгезии клеток.[33]
В качестве ещё одного фактора индукции апоптоза рассматривается атака инфицированных клеток цитотоксическими Т-лимфоцитами, которые, помимо активации Fas-рецептора, способны секретировать перфорин вблизи мембраны заражённой клетки. Перфорин, полимеризуясь, образует трансмембранные каналы, через которые внутрь клетки поступают лимфотоксин-альфа и смесь сериновых протеаз (гранзимов). Далее гранзим B активирует каспазу-3 и запускается каспазный каскад.[34]
Возможна инициация клеточной смерти при высвобождении лизосомальных протеаз — катепсинов. К примеру, каспаза-8 вызывает выход из лизосом активного катепсина B, который затем расщепляет регуляторный белок Bid[англ.]. В результате образуется активный белок t-Bid, активирующий в свою очередь проапоптозный белок Bax[англ.].[3]
Эффекторная фаза
В течение эффекторной фазы различные инициирующие пути конвертируются в один (или несколько) общий путь апоптоза.[17] Как правило, происходит активация каскада белков-эффекторов и регулирующих их белков-модуляторов.[1] Основными эффекторами апоптоза являются каспазы.[3] В процессе активации они запускают каспазный каскад - сложные цепочки взаимодействий инициирующих и эффекторных каспаз.
Каспазный каскад

Каспазы представляют собой цистеиновые протеазы, которые расщепляют аминокислотные последовательности после остатка аспарагиновой кислоты.[35] Каспазы образуются за счёт активации прокаспаз (молекулярная масса 32—56 кДа), в составе которых выделяют 3 домена: регуляторный N-концевой домен (продомен), большую (17—21 кДа) и малую (10—13 кДа) субъединицы.[3][36] Активация происходит путём протеолитического процессинга: все три домена расщепляются, отделяется продомен, а оставшиеся большая и малая субъединицы ассоциируются, образуя гетеродимер. Два гетеродимера в дальнейшем формируют тетрамер — полноценную каспазу с двумя каталитическими участками.[22]

один из которых инициирует, а другой подавляет апоптоз[22])
Каспазы обнаружены в большинстве живых организмов.[37] У млекопитающих идентифицировано 13 каспаз.[36] Часть из них в апоптозе не участвует (−1, −4, −5, −11, −13). Остальные каспазы, которые участвуют в апоптозе, разделяют на инициаторные (−2, −8, −9, −10, −12) и эффекторные (−3, −6, −7).[3] Инициаторные каспазы активируют эффекторные каспазы, которые в свою очередь провоцируют и непосредственно участвуют в трансформации клетки. В итоге морфологические и биохимические изменения приводят к гибели клетки по типу апоптоза.
Одна из основных функций эффекторных каспаз заключается в прямом и опосредованном разрушении клеточных структур. Гидролизу подвергаются белки ядерной ламины, разрушается цитоскелет, расщепляются белки, регулирующие клеточную адгезию. Другой важной функцией эффекторных каспаз является инактивация белков, блокирующих апоптоз. В частности расщепляется ингибитор DFF (англ. DNA fragmentation factor — «фактор фрагментации ДНК»), препятствующий активации апоптозной ДНКазы CAD (англ. caspase-activated DNase — «ДНКаза, активируемая каспазами»). Разрушению подвергаются и антиапоптозные белки семейства Bcl-2. Наконец, в результате действия эффекторных каспаз происходит диссоциация регуляторных и эффекторных доменов, участвующих в репарации ДНК, мРНК-сплайсинга и ДНК-репликации.[22][38]
Дополнительные эффекторы апоптоза
Помимо каспаз существуют и другие эффекторы апоптоза. Например, флавопротеин AIF, высвобождающийся из межмембранного пространства митохондрий, действует по независимому от каспаз пути. Попадая в клеточное ядро, AIF вызывает конденсацию хроматина и активирует эндонуклеазы, которые участвуют во фрагментации ДНК. На основании экспериментальных данных установлено, что апоптоз, протекающий в присутствии AIF, не предотвращается ингибитором каспаз (Z-VAD-fmk).[30] В качестве эффекторов апоптоза также рассматриваются кальпаины[39] — представители семейства цитозольных Ca2+-активируемых цистеиновых протеаз.[40] Их роль в апоптозе пока слабо охарактеризована.
Деградационная фаза
Итогом программируемой клеточной гибели вне зависимости от инициирующего воздействия является деградация клетки путём фрагментации на отдельные апоптотические тельца, ограниченные плазматической мембраной. Фрагменты погибшей клетки обычно очень быстро (в среднем за 90 минут[1]) фагоцитируются макрофагами либо соседними клетками, минуя развитие воспалительной реакции.
Морфологические изменения
Условно деградацию погибающей клетки можно разделить на три последовательных фазы: высвобождения, блеббинга и конденсации.[41] Деградация большинства клеток начинается с высвобождения прикреплений внеклеточного матрикса и реорганизации фокальной адгезии. Внутри погибающей клетки деполимеризуются микротрубочки цитоскелета. Внутриклеточные актиновые микрофиламенты реорганизуются в связанные с мембраной периферийные (кортикальные) кольцевые пучки. В итоге клетка приобретает округлую форму.[41] Следующая за высвобождением стадия блеббинга характеризуется сокращением периферийных актиновых колец. В результате сокращений клеточная мембрана образует вздутия, клетка как бы «кипит».[42] Процесс блеббинга энергозависим и требует большого количества АТФ.[43] Фаза блеббинга в нормальных условиях завершается примерно через час. В итоге клетка фрагментируется на маленькие апоптотические тела, либо целиком конденсируется, округляясь и уменьшаясь в размерах.[44]
Биохимические изменения
На молекулярном уровне одним из последствий апоптоза является фрагментация ДНК с участием нуклеаз. Изначально образуются крупные фрагменты с 30 000—700 000 пар оснований, которые в дальнейшем расщепляются в межнуклеосомной области на отрезки по 180—190 пар (180—200 пар[22]) оснований или кратные этим величинам.[18][45] Фрагментация ДНК является характерным, но не обязательным признаком апоптоза, так как существуют наблюдения, в ходе которых процесс фрагментации ядра (кариорексис) протекал без сопутствующей фрагментации ДНК.[22]
Ещё одним существенным последствием апоптоза является экспрессия на внешней стороне плазматической мембраны специфических молекулярных маркеров, распознаваемых фагоцитирующими клетками: тромбоспондина; фосфатидилсерина и других фосфолипидов, содержащих фосфосерин.[18][45] Маркеры, участвующие в фагоцитозе погибающей клетки, можно условно разделить на три группы: «ешь меня», «найди меня» и «не ешь меня».[46]
Критический сигнал «ешь меня» возникает при экстернализации фосфатидилсерина. Обычно он локализуется на внутреннем слое плазматической мембраны. Данное состояние обеспечивается АТФ-зависимой флиппазой, которая перемещает фосфолипид с внешнего на внутренний слой. Во время апоптоза фосфатидилсерин, наоборот, перемещается на внешний слой плазматической мембраны при участии каспаз. Там он ассоциируется с такими белками, как аннексин I и MFG-E8, которые участвуют во взаимодействии с рецепторами фагоцитирующих клеток.[47]
В процессе апоптоза также снижается интенсивность сигналов «не ешь меня» (CD31), которые в норме присутствуют у фагоцитов и у большинства здоровых клеток. Третья группа сигналов «найди меня» (лизофосфатидилхолин) продуцируется гибнущей клеткой для привлечения фагоцитов: эффекторные каспазы активируют фосфолипазу A, которая участвует в образовании лизофосфатидилхолина.[47]
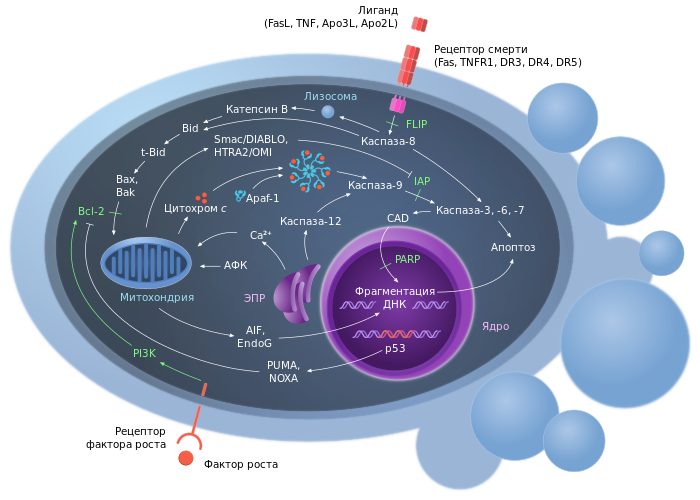
Регуляция апоптоза
Семейство белков Bcl-2
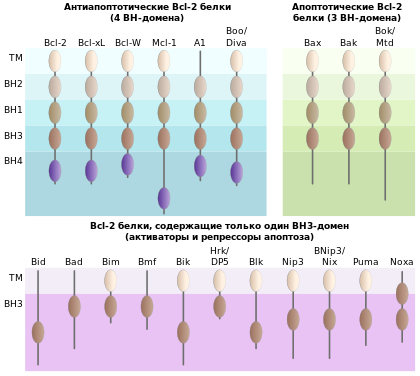
BH1—4 — гомологичные домены Bcl-2.
TM — трансмембранные домены
Белки семейства Bcl-2 являются основными регуляторами митохондриального пути апоптоза.[27][49] Они оказывают решающее воздействие на изменение проницаемости наружной мембраны митохондрий (MOMP). В семействе Bcl-2 различают проапоптотические и антиапоптотические белки. На основании структурных и функциональных различий выделяются три подсемейства белков Bcl-2[27]:
- Антиапоптотические Bcl-2-белки, содержащие 4 BH-домена[~ 2] (BH1—4): Bcl-2, Bcl-xL, Bcl-W, Mcl-1, A1, Boo/Diva;
- Проапоптотические Bcl-2-белки, содержащие 3 BH-домена (BH123): Вах, Bak, Bok/Mtd;
- Bcl-2-белки, содержащие только BH3-домен, которые могут исполнять роль активаторов или репрессоров апоптоза: Bid, Bad, Bim, Bmf, Bik, Hrk, Blk, Nip3, BNip3/Nix,Puma, Noxa.
Существенную роль в повышении MOMP играют апоптотические Bcl-2-белки — Bax и Bak. Они встраиваются в наружную мембрану митохондрий и олигомеризуются. При этом, вероятно, нарушается целостность внешней мембраны митохондрий, по неизвестному пока механизму.[27][50][51] Функционирование белков Bax и Bak зависит от их предварительной активации, например, белками Bid и Bim, которые относятся к подсемейству BH3-белков. С другой стороны активация и функционирование Bax и Bak может блокироваться антиапоптотическими белками семейства Bcl-2: Bcl-2, Bcl-xL, Mcl-1 и др. В свою очередь, антиапоптотические белки также могут блокироваться белками-репрессорами (например, Bad), относящимися к подсемейству BH3-белков. В итоге достигается комбинированная регуляция MOMP и, соответственно, апоптоза за счёт взаимодействия апоптотических, антиапоптотических, а также BH3-белков-активаторов и репрессоров. Регуляция функций BH3-белков осуществляется на уровне транскрипции, стабильности молекул, при взаимодействии с другими белками и при различных модификациях.[52]
Установлено также, что белок Bid является связующим звеном между рецептор-зависимым и митохондриальным путями апоптоза. Активированная через рецепторы клеточной гибели инициирующая каспаза-8 способна активировать белок Bid. Далее Bid участвует в активации белков Bax и Bak, которые запускают митохондриальный путь апоптоза.[53]
Дополнительно имеются сведения о том, что белки семейства Bcl-2 могут выступать в роли адаптеров, связывающихся с белками, участвующими в процессе апоптоза.[54] Например, Bcl-xL может ингибировать соединение APAF-1 с прокаспазой-9, предотвращая активацию каспазы-9.[55]
Ингибиторы белков апоптоза
Ингибиторы белков апоптоза (англ. inhibitors of apoptosis proteins, IAPs) впервые были обнаружены у бакуловирусов. Вслед за этим открытием гомологи IAP были выявлены у всех эукариот, от дрожжей до млекопитающих. В структуре IAP выделяют от одного до трёх 70-аминокислотных N-концевых BIR-доменов (от англ. baculoviruses inhibitor of apoptosis repeat domains). К тому же X-хромосомный XIAP и клеточный cIAP1,2 (англ. cellular IAP) содержат С-концевой RING-домен (от англ. really interesting new gene)[3].
Основная роль ингибиторов белков апоптоза заключается в подавлении функционирования каспаз −3, −7, −9. При этом BIR-домены связывают активные сайты каспаз, а RING-домены участвуют в деградации каспаз за счёт убиквитин-лигазной активности. Действие IAP подавляется регуляторами Smac/DIABLO и Omi/HtrA2, высвобождающимися из межмембранного пространства митохондрий. Помимо этого каспазы −3 и −7 при сверхэкспрессии способны самостоятельно расщеплять XIAP[3].
FLIP (англ. FLICE-inhibitory protein — «белок, ингибирующий FLICE») — внутриклеточный ингибитор каспазы-8, блокирующий передачу сигнала апоптоза через рецепторы смерти[3]. Роль FLIP является противоречивой, так как его сверхэкспрессия или ингибирует, или активирует апоптоз.[56]
Альтернативные пути передачи сигнала от рецепторов смерти
Активация рецепторов смерти TNFR1 или DR3 ведёт к равновероятному запуску двух альтернативных путей, один из которых оканчивается апоптозом, а другой препятствует индукции апоптоза. Дело в том, что адаптер TRADD, связанный с рецепторами TNFR1 и DR3, наряду с активацией прокаспазы-8, участвует в активации ядерных факторов транскрипции NF-kB (от англ. nuclear factor kappa B — «ядерный фактор каппа B») и JNK/AP-1 (JNK, Jun-N-концевая киназа, является компонентом митоген-активируемого киназного пути, ведущего к активации фактора транскрипции AP-1). Факторы NF-kB и JNK/AP-1 в свою очередь контролируют синтез белковых регуляторов, которые блокируют TNF- или Apo3L-индуцированную активацию каспазы-8, вследствие чего подавляется опосредованный рецепторами апоптоз.[22]
Дополнительными регуляторами в рецептор-зависимом сигнальном пути апоптоза являются «обманные» DcR-рецепторы, которые конкурируют с DR4 и DR5 в связывании лиганда Apo2L.[57] DcR1-рецептор — клеточный поверхностный белок, у которого отсутствует цитоплазматический хвост.[58] DcR2 — рецептор со значительно укороченным цитоплазматическим доменом смерти.[59] Оба рецептора, DcR1 и DcR2, благодаря структурному сходству с DR4 и DR5, подавляют активацию апоптоза лигандом Apo2L.
Белок p53
В нормальных клетках белок p53, как правило, находится в неактивной, латентной форме. Активация p53 происходит в ответ на повреждения ДНК, вызванные ультрафиолетовым или гамма-излучением, гиперэкспрессией онкогенов, вирусной инфекцией, оксидативным стрессом, гипо- и гипертермией и др.[60] Активированный p53 координирует процесс репарации ДНК, а также регулирует транскрипцию ряда генов-активаторов апоптоза в случае необратимых повреждений ДНК или нарушений регуляции клеточного цикла. К тому же имеются указания на то, что p53 принимает участие в запуске апоптоза путём стимуляции рецепторов смерти, путём взаимодействия с промотором апоптоза — Bax, путём активации p53-зависимого модулятора апоптоза PUMA (англ. p53 upregulated modulator of apoptosis), который блокирует действие Bcl-2[3]. Повышение уровня p53 в ответ на повреждения ДНК вызывает апоптоз, например, в клетках кожи, в тимоцитах, в клетках кишечного эпителия.[61]
Роль апоптоза в многоклеточном организме
Редукция хвоста у головастика при превращении в лягушку


Клеточный гомеостаз и морфогенез
Одной из главных функций апоптоза в многоклеточном организме является поддержание клеточного гомеостаза, то есть постоянства клеточной популяции. При этом обеспечивается правильное соотношение численности клеток различных типов, селекция разновидностей клеток внутри популяции, удаление генетически дефектных клеток.[62] Во взрослом организме программируемая клеточная гибель, уравновешивая митотическое деление, обеспечивает обновление тканей путём поддержания сбалансированной численности клеток.[18] В качестве примера, иллюстрирующего роль апоптоза в поддержании численности отдельных клеточных популяций, может служить увеличение численности эндотелиальных клеток и размера сосудов у мышей с прицельной инактивацией гена Braf, контролирующего апоптоз эндотелиальных клеток.[62]

Велика роль апоптоза в формообразовательных процессах, в дифференциации тканей и отдельных частей органов. У животных роль апоптоза в морфогенезе отдельных органов или их частей наиболее отчётливо прослеживается в процессе эмбриогенеза. К примеру, утрата хвоста зародышами амфибий или атрофия у них гипохорды объясняются массовым апоптозом целых клеточных популяций.[63] С другой стороны, уже во взрослом организме, атрофия гормонально-зависимых тканей в условиях снижения концентрации соответствующих гормонов также обусловлена апоптозом. Например, процессы такого рода периодически протекают в женских половых органах в течение менструального цикла, или же в предстательной железе при снижении концентрации андрогенов.[63] В растительном организме апоптоз не менее интегрирован в процессы морфогенеза и дифференциации, чем у животных. Программируемая клеточная смерть у растений обеспечивает ксилогенез и флоэмогенез, формообразование листьев, аэренхимогенез, образование корневого чехлика, опадание листьев и созревших плодов, прорастание пыльцевой трубки и др.[22]
Роль апоптоза в иммунных процессах
В иммунной системе животных программируемая клеточная смерть задействована в обеспечении целого ряда жизненно важных функций иммунитета. Процесс апоптоза заложен в основу позитивной и негативной селекции T- и B-лимфоцитов, обеспечивая выживание антигенспецифичных клонов и последующую выбраковку аутореактивных лимфоцитов.[18] На обоих этапах селекции клетки, не прошедшие отбор, погибают в результате апоптоза.[64] Немаловажна роль программируемой клеточной смерти в реализации эффекторной функции цитотоксических Т-клеток и НК-клеток — и те и другие способны инъецировать внутрь клеток-мишеней сериновые протеазы (гранзимы), которые запускают механизм апоптоза. Помимо этого цитотоксические Т-лимфоциты способны инициировать клеточную гибель посредством активации рецепторов смерти на поверхности клеток-мишеней.[65] Ещё одной установленной функцией апоптоза в рамках иммунной системы является изоляция «иммунологически привилегированных» зон (например, внутренней среды глаза или семенников). При этом клетки, выполняющие барьерную функцию, инициируют рецептор-зависимый апоптоз эффекторных Т-лимфоцитов, мигрирующих сквозь «барьерные» ткани.[66]
Растительная иммунная реакция на патогенные вирусы, бактерии, грибы и нематоды протекает в форме гиперчувствительного ответа — программируемой гибели инфицированных клеток, а также клеток, локализованных вблизи очага инфекции. Таким образом, наряду с защитным процессом синтеза фитоалексинов и гидролитических ферментов, образуется барьерная зона мёртвых обезвоженных клеток, препятствующих распространению патогена.[22]
Роль апоптоза в процессах старения
Предположение о роли апоптотической гибели в процессах старения было высказано ещё в 1982 году. Со временем выяснилось, что различные виды возрастзависимой дисрегуляции апоптоза присущи многим типам клеток. Например, в стареющем организме повышается чувствительность к индукции апоптоза для следующих типов клеток: гепатоцитов, кардиомиоцитов, макрофагов, мегакариоцитов, нейронов, ооцитов, спленоцитов, T-лимфоцитов, хондроцитов, эндотелиоцитов. Но в то же время для фибробластов наблюдается обратная тенденция к снижению чувствительности к апоптозу, а для кератиноцитов данная чувствительность не изменяется.[67]
К настоящему времени имеются как минимум две точки зрения на связь апоптоза с процессами старения. Согласно одной из версий нормальные (гомеостатические) апоптотические процессы могут участвовать в развитии возрастных патологий и фенотипов старения.[68] К примеру, с апоптотической гибелью постмитотических клеток (кардиомиоцитов, нейронов) связаны процессы старения сердечной мышцы или развитие возрастных нейродегенеративных патологий.[69] Старение иммунной системы также связывают с программируемой гибелью различных типов лейкоцитов в результате возрастных изменений в соотношении про- и антиапоптозных факторов. Возрастная хрящевая дегенерация коррелирует с повышением уровня апоптоза хондроцитов в суставных хрящах у мышей и крыс, а также в межпозвоночных дисках при старении у человека.[70] Согласно другой точке зрения накопление стареющих клеток в тканях объясняется возрастной резистентностью к апоптозу.[68] В качестве примера рассматривается устойчивость стареющих фибробластов к апоптозу, приводящая в итоге к преждевременному старению нормальных фибробластов и, возможно, к нарушению функций соединительной ткани.[71]
Патология, обусловленная нарушениями апоптоза

Организмы с обширными дефектами, обусловленными нарушениями апоптоза, погибают ещё на ранних стадиях онтогенеза. Регистрируются лишь локальные дефекты, развившиеся в ходе эмбрионального развития, или же дефекты с ограниченными фенотипическими проявлениями, развившиеся уже во взрослых организмах. Патологические процессы развиваются в случае подавления или усиления апоптоза. При недостаточности апоптоза прогрессируют аутоиммунные процессы и злокачественные новообразования. При усилении апоптоза возникают аплазии и дегенеративные процессы, а также некоторые уродства с дефектами тканей.[72]
Патология, связанная с ослаблением апоптоза
Вирусные заболевания
В норме инфицированные клетки погибают в результате активации рецептор-зависимого апоптоза, дабы предотвратить распространение вируса. Однако некоторые вирусы способны нарушать нормальную регуляцию механизма программируемой клеточной гибели, или даже активно предотвращать апоптоз. Вирусная блокада клеточной гибели может быть основана на синтезе IAP, гомологов белка Bcl-2, а также других ингибиторов апоптоза.[73]
Бакуловирусы насекомых блокируют апоптоз за счёт экспрессии IAP, блокирующих инициаторную и эффекторные каспазы. К тому же бакуловирусы экспрессируют белок p35, который связывает и ингибирует активные каспазы. В результате жизнеспособность заражённой клетки поддерживается до образования достаточного количества вирусных частиц.[74]
Вирусы позвоночных способны блокировать апоптоз путём синтеза антиапоптотических белков семейства Bcl-2, например E1B19K и белок BHRF вируса Эпштейна — Барр. К тому же вирусы позвоночных часто функционируют, предотвращая апоптоз, запускаемый клетками иммунной системы. Например, вирус ветряной оспы продуцирует серпины, блокирующие гранзим B и каспазу-8. Тем самым, инфицированная клетка избегает воздействия со стороны цитотоксических лимфоцитов, а также избегает апоптоза. Другой пример: вирус герпеса продуцирует белок v-FLIP, который блокирует апоптоз, реализуемый с участием рецепторов клеточной гибели.[74]
Опухолевые заболевания
Вторую группу заболеваний, связанных с ослаблением апоптоза, составляют злокачественные опухоли. В качестве основной причины данной патологии рассматривают соматические мутации гена, кодирующего белок p53. Порядка 50 %[74] (70 %[75]) трансформированных клеток экспрессируют мутантную форму p53. Механизм подавления программируемой клеточной гибели также может быть связан с повышенной экспрессией или мутацией гена Bcl-2.[76] Например, установлен факт рекомбинации гена Bcl-2 при лимфоме Беркитта и некоторых формах фолликулярных лимфом.[75]
Аутоиммунные заболевания
Основным признаком аутоиммунной патологии является иммунная реакция против собственных клеток и тканей организма, причиной чему может быть сбой в программе негативной селекции T-лимфоцитов. Нарушение T-клеточного апоптоза позволяет выжить аутореактивным клонам T-лимфоцитов. Вдобавок нарушается формирование полного состава апоптозных аутоантигенов (свойственных организму белков-участников апоптоза), к которым должна развиться толерантность. Как следствие, малые по интенсивности проапоптозные воздействия приводят к повышению уровня аутоантигенов, участвующих в апоптозе, что в свою очередь влечёт за собой проявление клинических признаков аутоиммунной патологии. Примером могут служить аутоиммунные дерматиты, прогрессирующие при воздействии солнечных лучей или при снижении температуры окружающей среды.[76]
Патология, связанная с усилением апоптоза
Одной из групп заболеваний, связанных с усилением апоптоза, являются патологии системы крови. Чаще всего патологические процессы развиваются в результате гибели костномозговых клеток-предшественников посредством апоптоза. Причиной их гибели является недостаточность факторов выживания. Данный тип патологии приводит к развитию апластической анемии; анемии при дефиците железа, фолатов, витамина B12; талассемии; тромбоцитопении; лимфопении; нейтропении; панцитопении. Повышенная готовность к развитию апоптоза Т-лимфоцитов обнаружена при мультицентрической болезни Кастелмана.[72]
Прогрессия некоторых инфекционных заболеваний может быть связана не только с подавлением, но и наоборот, с усилением апоптоза. Индукторами программируемой клеточной гибели при этом служат бактериальные эндо- и экзотоксины. Массовый апоптоз развивается при сепсисе. Гибель лимфоцитов путём апоптоза находится в положительной корреляции с быстрой прогрессией СПИДа.[72]
Отдельную группу патологии составляют заболевания нервной системы, обусловленные атрофией определённых участков нервной ткани в результате апоптоза. Примерами таких заболеваний могут служить боковой амиотрофический склероз, болезнь Альцгеймера, спинальная мышечная атрофия и др.[72]
Апоптоз является преобладающей формой гибели миоцитов в ранний период развития инфаркта.[72] На основе экспериментальных данных было выявлено, что программируемая гибель кардиомиоцитов может быть обусловлена гипоксией, ишемией, перегрузкой клетки кальцием, воспалением, токсинами.[77] В процессе токсического (в том числе и алкогольного) гепатита основная роль также отводится апоптозу.[72]
Ряд патологических процессов, обусловленных усилением апоптоза, индуцируется внешними апоптогенными факторами. Апоптоз прогрессирует под воздействием ионизирующей радиации. При этом преимущественно гибнут лимфоидные клетки и развивается иммунная недостаточность. Аналогичный эффект дают многие химиотерапевтические препараты, используемые при лечении опухолей, а также гормоны, применяемые при лечении различных заболеваний.[72]
Происхождение и эволюция апоптоза
Предположительно, апоптоз эволюционно возник у прокариот и закрепился у одноклеточных эукариот, в качестве механизма противовирусной защиты популяций. В дальнейшем, с появлением многоклеточных организмов, механизм программируемой клеточной смерти совершенствовался и был приспособлен, наряду с защитой от патогенов, для реализации важных жизненных функций: дифференцировки клеток и тканей при эмбриогенезе и постэмбриональном развитии; элиминирования клеток иммунной системы, невостребованных, состарившихся клеток либо клеток, подвергшихся воздействию мутагенных факторов.[22][78]
Апоптоз у прокариот
Предполагается, что механизмы программируемой клеточной смерти возникли ещё у прокариот, подтверждением чему служит целый ряд экспериментальных данных. В частности, выявлена роль апоптоза в противовирусной защите бактериальных популяций. К примеру, некоторые штаммы E. coli несут гены, вызывающие гибель клетки после внедрения фага T4. При этом вирусные белки активируют в заражённых клетках протеазы, которые инактивируют и расщепляют бактериальный фактор трансляции (EF-Tu), что приводит к гибели заражённой бактерии[3][22].
Прокариотическим аналогом апоптоза также считается гибель части бактерий в условиях стазиса — остановки роста бактериальной популяции (при исчерпании питательного субстрата или под влиянием стрессорных факторов). Например, голодающая популяция E. coli разделяется на две субпопуляции, одна из которых погибает и подвергается автолизу. Выжившая популяция в итоге использует продукты автолиза в качестве питательного субстрата и продолжает расти. Механизм программируемой клеточной гибели в данном случае основан на формировании модулей зависимости. Модуль зависимости представляет собой неактивный комплекс из стабильного цитотоксического белка и его нестабильного супрессора. В условиях голода прекращается синтез обоих белков. В результате нестабильный супрессор разрушается, а цитотоксический белок вызывает гибель и автолиз.[22]
Имеются данные о функциональной роли программируемой клеточной смерти в процессах развития и морфогенеза прокариот. Так, в условиях гибели значительной части клеточной популяции протекает образование плодового тела и споруляция у Myxococcus xanthus. Другим примером может служить процесс спорообразования у бациллы Bacillus subtilis: материнская вегетативная клетка погибает и активно лизируется при высвобождении споры.[3][22]
Апоптоз у одноклеточных эукариот
Примеры программируемой клеточной гибели описаны и для представителей одноклеточных эукариот, принадлежащих к различным таксонам. Причём во многих исследованиях установлен факт участия цистеиновых протеаз и митохондрий в гибели клеток, что может указывать на очень древнее происхождение и относительную консервативность механизмов апоптоза. Основными маркерами апоптоза у одноклеточных эукариот, как и у большинства эукариот вообще, являются: фрагментация ДНК и последующий распад клетки на отдельные апоптозные тельца.[3]
Предполагается, что отдельные механизмы и компоненты апоптоза возникли постепенно в процессе эволюции. Одними из самых ранних эволюционных приобретений считаются ингибиторы апоптоза, которые встречаются практически у всех эукариот. Вероятно, ингибиторы имеют вирусное происхождение, а их изначальная функция сводилась к предотвращению апоптоза и продлению жизни инфицированной клетки. Ещё одним общим для подавляющего большинства эукариот[~ 3] эволюционным приобретением является митохондриальный путь активации апоптоза, в котором участвуют высвобождаемые из межмембранного пространства митохондрий цитохром c и AIF (или их гомологи).[3]
Подобные каспазам белки впервые появились у мезокариотических водорослей — динофлагеллят. Причём инициаторные каспазы возникли, предположительно, раньше, чем эффекторные.[~ 4] Рецепторы смерти, вероятно, впервые возникли у относительно высокоорганизованных простейших — дрожжей и инфузорий. Белки, способные взаимодействовать с белками семейства Bcl-2, вероятно, появились в процессе филогенеза у гетеротрофных жгутиконосцев, хотя идентифицированы они только у дрожжей.[3]
Одной из основных функций апоптоза у одноклеточных эукариот является уничтожение мутантных или инфицированных клеток. Механизмы программируемой клеточной гибели могут быть сопряжены с процессами дифференцировки. Примерами чему служат избирательная гибель ядра у конъюгирующих инфузорий или массовая гибель эпимастигот с появлением трипомастигот в процессе жизненного цикла паразитического жгутиконосца Trypanosoma cruzi.[22] Апоптоз также интегрирован в процессы морфогенеза при образовании плодовых тел у миксобактерий и слизевиков. В дрожжевых популяциях стареющие и повреждённые клетки погибают путём апоптоза в условиях недостатка питательного субстрата с целью обеспечения питания молодых и здоровых особей.[3]
Апоптоз у многоклеточных эукариот
Принципиально апоптоз у многоклеточных эукариот сходен с программируемой клеточной гибелью у одноклеточных эукариот. На протяжении всего эволюционного процесса прослеживается общность основных функций апоптоза, сводящихся к удалению дефектных клеток и участию в процессах дифференцировки и морфогенеза. В различных литературных и электронных источниках постулируется эволюционная консервативность генетического механизма апоптоза.[3][16][79] В частности, подобные выводы делаются на основании выявленной генетической и функциональной гомологии процессов апоптоза у нематод Caenorhabditis elegans и млекопитающих,[3] или же у растений и животных.[16]
Апоптоз можно рассматривать как небольшое видоизменение митоза, как эволюционную надстройку над делением. Внешне апоптоз также напоминает процесс деления, только в отличие от последнего он заканчивается не образованием двух полноценных дочерних клеток, а несколько большим количеством апоптозных микротелец, также несущих в себе генетический материал и удобных для фагоцитирования. При таком подходе к апоптозу становится понятным тот факт, что широкий набор генетических маркеров пролиферации являются и маркерами апоптозной гибели[80].
Апоптоз у Caenorhabditis elegans

Нематода C. elegans была одним из первых модельных организмов, на примере которого изучался процесс апоптоза. В процессе развития C. elegans путём апоптоза погибает 131 из 1090 соматических клеток взрослого организма нематоды. Механизм программируемой клеточной гибели нематод напоминает митохондриальный путь апоптоза позвоночных, но в то же время имеет и существенные отличия.[82]
В реализации программированной клеточной гибели по пути апоптоза у нематоды участвует одна каспаза, являющаяся продуктом гена ced-3 (англ. C. elegans death gene, CED-3). Каспаза CED-3 содержит CARD-домен. Активация каспазы CED-3 осуществляется при взаимодействии её CARD-домена с CARD-доменом адаптерной молекулы CED-4. Адаптерная молекула CED-4 по структуре гомологична APAF-1 и аналогичным образом олигомеризуется перед активацией каспазы CED-3. В свою очередь, каспаза CED-3 содержит CARD-домен и активируется за счёт димеризации, также как каспаза-9 у позвоночных.[82]
Регуляция апоптоза у нематоды обеспечивается за счёт белков CED-9 и EGL-1. CED-9 является гомологом антиапоптотического белка Bcl-2 позвоночных, но функционально отличается от него. CED-9, вероятно, блокирует адаптерный белок CED-4 за счёт прямого связывания. Связывание CED-9 и CED-4 нарушается белком BH3-семейства — EGL-1 (egg laying deficient). В результате высвобождения адаптерного белка CED-4 активируется каспаза CED-3, что в итоге приводит к клеточной гибели.[81]
Апоптоз у Drosophila melanogaster

Как и у нематод, апоптоз у насекомых схож с митохондриальным путём этого процесса у позвоночных, но в то же время характеризуется существенными различиями. У Drosophila melanogaster апоптоз наступает в процессе развития (под действием гормона экдизона) или при клеточном стрессе (например, при повреждении ДНК).[81]
Апоптоз у насекомых протекает с участием двух эффекторных каспаз: DRICE и DCP-1. Активацию эффекторных каспаз обеспечивает инициаторная каспаза DRONC в комплексе с белком ARK (англ. APAF-1 related killer), который представляет собой гомолог APAF-1. Однако пути активации и функциональная активность белка ARK остаются пока малоизученными.[81]
Важным фактором, обеспечивающим регуляцию и протекание апоптоза у Drosophila melanogaster является белковый ингибитор апоптоза IAP, и в особенности, DIAP 1. Ингибиторы апоптоза блокируют инициаторную каспазу DRONC и эффекторные каспазы DRICE и DCP-1, предотвращая гибель клетки. В связи с этим для запуска апоптоза у Drosophila экспрессируются один или несколько белков, являющихся антагонистами IAP: Reaper, Hid, Grim, Sickle. Эти белки блокируют действие IAP, вследствие чего высвобождаются инициаторные и эффекторные каспазы, участвующие в апоптозе.[81]
Апоптоз у растений
Программируемая клеточная смерть (ПКС) растений имеет морфологические и биохимические сходства с апоптозом животных. Однако особенности строения и функционирования растительной клетки обуславливают и ряд отличий. Например, наличие клеточной стенки препятствует фагоцитозу, поэтому в растительных клетках продукты апоптоза ликвидируются за счёт аутофагии и автолиза. А имеющиеся в клетках растений вакуоли при этом используются в качестве гидролитических отсеков.[83]
Программируемая клеточная смерть играет ключевую роль во многих вегетативных и репродуктивных фазах развития растений, включая старение листьев, ксилогенез, отмирание лепестков после оплодотворения, постэмбриональный распад алейроновых слоёв, развитие корневого чехлика, соматический и зиготический эмбриогенез, определение пола. ПКС в растениях может развиваться в ответ на биотические и абиотические раздражители. Авирулентные инфекции, как правило, характеризуются локальной гибелью клеток, известной как гиперчувствительный ответ, приводящий к образованию некротических поражений вокруг инфицированных областей. В ответ на абиотический стресс также может происходить активация ПКС. Наилучшим примером служит образование аэренхимы в условиях низкой концентрации кислорода, при которой кортикальные клетки корня подвергаются индуцированной гибели и тем самым формируют крупные воздушные резервуары, обеспечивая поступление воздуха из верхней части растения. ПКС у растений также может происходить при воздействии высоких температур.[84]
| Животные | Растения |
|---|---|
| ДНК фрагментируется на участки длиной примерно 180 пар оснований. | Длина фрагментов ДНК варьирует от 140 до 50 000 пар нуклеотидов. Фрагментация ДНК не наблюдается в трахеидах, древесинных и лубяных волокнах. |
| Ca2+-зависимые эндонуклеазы участвуют во фрагментации ДНК. Почти во всех случаях (за исключением одного случая у C. elegans) нуклеазы является продуктом самой погибающей клетки. | Нуклеазы встречаются в некоторых погибающих клетках растений, но пока не получены прямые доказательства их участия в программируемой клеточной смерти. Растительные нуклеазы могут быть Ca2+ или Zn2+-зависимые. Некоторые нуклеазы активируются как Ca2+, так и Zn2+. Нуклеазы могут синтезироваться самой погибающей клеткой или транспортироваться из соседних. |
| В процессе апоптоза происходит накопление фосфатидилсерина на внешней стороне клеточной мембраны. Наличие фосфатидилсерина может быть обнаружено за счёт связывания аннексина V. | Наличие фосфатидилсерина (за счёт связывания аннексина V) пока обнаружено только в протопласте табака, подвергнутого абиотическому стрессу. |
| Конденсация, усадка и фрагментация цитоплазмы всегда заметны. | У растений отмечается конденсация и усадка цитоплазмы, но без фрагментации. |
| Клетки сжимаются, сокращаются в размерах. | Клетки большинства типов тканей сжимаются, «усыхают» в процессе гибели. Исключение составляют трахеиды, древесинные и лубяные волокна. |
| Процесс апоптоза представляет собой комплекс хорошо организованных и отлаженных механизмов с участием эффекторов, адаптеров, регуляторов и сигналов. | Механизм программируемой клеточной гибели пока остаётся недостаточно исследованным. Выявлены эффекторы, сигнальные и некоторые регуляторные молекулы. Не известны адаптеры. |
| Экспрессированные и активированные эффекторные каспазы (цистеиновые протеазы) и гранзимы избирательно расщепляют целевые белки в области аспартатных остатков. | Экспрессия цистеиновых протеаз наблюдается в некоторых случаях, но не исключительно в процессе клеточной гибели. Субстраты для некоторых цистеиновых протеаз не определены. Специфика каспаз также не известна. Гомология каспазам животных очень низкая. |
| Погибающие клетки захватываются и элиминируются путём фагоцитоза соседними клетками или макрофагами. | После гибели растительной клетки сохраняется её клеточная стенка. Трахеиды, древесинные и лубяные волокна вообще начинают функционировать только после отмирания клеточного содержимого. Цитоплазма гибнущей клетки почти всегда элиминируется путём аутофагии в вакуолях, и, вероятно, в лизосомах. |
| Антиапоптозный белок Bcl-xL подавляет апоптоз, по крайней мере, в некоторых клетках. | Bcl-xL не подавляет апоптоз, связанный с гиперчувствительным ответом. |
| Активные формы кислорода, такие как O2 и H2O2, могут выступать в качестве сигнальных молекул, активирующих апоптоз. | O2 и H2O2 вовлечены в процесс клеточной гибели, особенно при гиперчувствительном ответе на биотический или абиотический стресс. Однако окончательных доказательств их участия в процессе гибели пока нет. |
| Увеличение концентрации Ca2+ в цитозоле может запустить апоптоз путём активирации эндонуклеаз и каспаз. | Увеличение концентрации Ca2+ в цитозоле может запустить ПКС путём активирации эндонуклеаз. Данные об Ca2+-зависимой активации протеинкиназ не получены. |
| Достоверно установлена роль митохондрий в процессе апоптоза. | Роль (если таковая имеется) митохондрий в ПКС является обоснованной. Существуют один или два доклада о причастности митохондрий к ПКС. |
| Клеточная мембрана подвергается блеббингу (пузырению). | Нет данных о блеббинге (пузырении) клеточной мембраны. |
| Сохраняется обычный уровень фосфорилирования/дефосфорилирования белков. | Фосфорилирование/дефосфорилирование белков зафиксировано только в клетках подвергнутых гипоксии и вовлечённых в гиперчувстительный ответ, а также в алейроновых клетках. |
| Исключение факторов роста способствует гибели клеток. | Существуют противоречивые данные о влиянии отсутствия факторов роста на гибель клеток. |
| Наблюдается конденсация хроматина в процессе апоптоза. | Конденсация хроматина при ПКС наблюдается не во всех типах клеток. |
| Расщепление и фрагментация ДНК наблюдается в опытах с применением ДНК-электрофореза с окрашиванием TUNEL-методом. Расщепление ДНК происходит в линкерных участках между нуклеосомами, и приводит к образованию олигонуклеосомных фрагментов. | Не во всех типах клеток расщепление и фрагментация ДНК наблюдается в опытах с применением ДНК-электрофореза с окрашиванием TUNEL-методом. |
| Типичные апоптозные тельца содержат часть цитоплазмы клетки и фрагменты ДНК. | Нет данных о формировании апоптозных тел. |
| Стрессовые белки не синтезируются в процессе клеточной гибели. | Стрессовые белки, такие как гидроксипролин, глицин, арабиногалактан, часто синтезируются и становятся неотъемлемыми компонентами клеточной стенки в некоторых типах погибающих клеток. |
Другие формы гибели клетки
В современной классификации программируемой клеточной смерти различают как минимум четыре основные формы программируемой клеточной смерти (ПКС):
- апоптоз — «ПКС I типа»
- аутофагия — «ПКС II типа»
- некроз — «ПКС III типа» (может использоваться синоним «онкоз»).
- аноикис — «ПКС IV типа» — смерть клетки, вызванная отделением её от окружающего внеклеточного матрикса[86].
- некроптоз — один из видов программируемого некроза[87].
- пироптоз — другой вид программируемого некроза.
- ферроптоз — тип программируемой окислительной некротической гибели клетки, характерной особенностью которого является железо-зависимое перекисное окисление липидов.
В качестве отдельных форм гибели клетки также рассматриваются митотическая катастрофа, сенессенс (клеточное старение), параптоз.[39][88]
См. также
Примечания
- Комментарии
- ↑ Лиганд – молекула, взаимодействующая с клеточным рецептором.
- ↑ От англ. Bcl-2 Homology — «гомологичный домен Bcl-2»
- ↑ Исключением может служить лишённое митохондрий простейшее Trichomonas vaginalis.[3]
- ↑ Эффекторные каспазы появляются только у примитивных многоклеточных — кишечнополостных.[3]
- Источники
- ↑ 1 2 3 4 Сербин М. Е., Щербак Е. В. Апоптоз и его молекулярные эффекторы // Актуальные проблемы биологии, медицины и экологии : Сборник / под редакцией проф., д. м. н. Н. Н. Ильинских. — Томск: Сибирский государственный медицинский университет, 2004. — Вып. 1. ()
- ↑ Кузнецов, Мушкамбаров, 2007, с. 83.
- ↑ 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 Гордеева и др., 2004, с. 1301—1313.
- ↑ 1 2 3 4 5 6 Michael G E O'Rourke and Kay A O Ellem. John Kerr and apoptosis (англ.). Medical Journal of Australia (2000). Архивировано 23 августа 2011 года.
- ↑ Chen, Lai (eds.), 2009, p. 27.
- ↑ 1 2 3 Banfalvi, 2009, p. 207.
- ↑ 1 2 Lockshin, Zakeri (eds.), 2004, p. 12.
- ↑ 1 2 Banfalvi, 2009, p. 203.
- ↑ 1 2 Vaux, 2002, p. 349.
- ↑ J. F. R. Kerr, A. H. Wyllie, A. R. Currie. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics (англ.). British Journal of Cancer (1972). Архивировано 1 ноября 2020 года.
- ↑ H. Robert Horvitz. Worms, life and death. Nobel lecture (англ.). nobelprize.org (8 декабря 2002). Архивировано 23 августа 2011 года.
- ↑ Banfalvi, 2009, p. 206.
- ↑ 1 2 Banfalvi, 2009, p. 204.
- ↑ Барышников, Шишкин, 2002, с. 15.
- ↑ Banfalvi, 2009, p. 205.
- ↑ 1 2 3 Ванюшин, 2001, с. 5.
- ↑ 1 2 3 Барышников, Шишкин, 2002, с. 13.
- ↑ 1 2 3 4 5 Апоптоз: введение. Сайт humbio.ru. Архивировано 23 августа 2011 года.
- ↑ 1 2 3 Барышников, Шишкин, 2002, с. 38.
- ↑ Льюин и др., 2011, с. 613.
- ↑ Передача сигнала включения апоптоза. Сайт humbio.ru. Архивировано 23 августа 2011 года.
- ↑ 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 Самуилов В. Д., Олескин А. В., Лагунова Е. М. Программируемая клеточная смерть. Кафедра физиологии микроорганизмов биологического факультета МГУ им. М. В. Ломоносова (6 июня 2001). Архивировано 26 декабря 2002 года.
- ↑ Прокаспаза 8 (FLICE/MACH/Mch5): активация. Сайт humbio.ru. Архивировано 23 августа 2011 года.
- ↑ 1 2 Alberts B. at al., 2008, p. 1122.
- ↑ 1 2 Льюин и др., 2011, с. 619.
- ↑ Модель активации каспаз митохондриями. Сайт humbio.ru. Архивировано 23 августа 2011 года.
- ↑ 1 2 3 4 Льюин и др., 2011, с. 620.
- ↑ 1 2 PT поры во внутренней мембране, разрушение внешней мембраны и апоптоз. Сайт humbio.ru. Архивировано 23 августа 2011 года.
- ↑ Льюин и др., 2011, с. 624.
- ↑ 1 2 AIF: фактор, индуцирующий апоптоз по независимому от каспаз пути. Сайт humbio.ru. Архивировано 23 августа 2011 года.
- ↑ Апоптоз с участием рецепторного и митохондриального механизмов. Сайт humbio.ru. Архивировано 23 августа 2011 года.
- ↑ Апоптоз с участием эндоплазматического ретикулума. Сайт humbio.ru. Архивировано 23 августа 2011 года.
- ↑ Апоптоз, вызванный нарушением адгезии клеток. Сайт humbio.ru. Архивировано 23 августа 2011 года.
- ↑ Апоптоз, вызванный цитотоксическими лимфоцитами. Сайт humbio.ru. Архивировано 23 августа 2011 года.
- ↑ Барышников, Шишкин, 2002, с. 41.
- ↑ 1 2 Барышников, Шишкин, 2002, с. 42.
- ↑ Предполагаемые функции и структура каспаз. Сайт humbio.ru. Архивировано 23 августа 2011 года.
- ↑ Барышников, Шишкин, 2002, с. 46.
- ↑ 1 2 Linda E. Bröker, Frank A. E. Kruyt and Giuseppe Giaccone. Cell Death Independent of Caspases: A Review (англ.). American Association for Cancer Research (2 мая 2005). Архивировано 3 февраля 2012 года.
- ↑ Кальпаин. Сайт humbio.ru. Архивировано 23 августа 2011 года.
- ↑ 1 2 Апоптоз: фаза экзекуции внеядерная: введение. Сайт humbio.ru. Архивировано 23 августа 2011 года.
- ↑ Апоптоз: фаза экзекуции: фаза высвобождения: блеббинг: введение. Сайт humbio.ru. Архивировано 23 августа 2011 года.
- ↑ Апоптоз: фаза экзекуции: фаза блеббинга: ATP. Сайт humbio.ru. Архивировано 23 августа 2011 года.
- ↑ Апоптоз: фаза экзекуции: фаза конденсации: введение. Сайт humbio.ru. Архивировано 23 августа 2011 года.
- ↑ 1 2 Барышников, Шишкин, 2002, с. 12.
- ↑ 1 2 Льюин и др., 2011, с. 631.
- ↑ 1 2 Льюин и др., 2011, с. 633.
- ↑ Льюин и др., 2011, с. 621.
- ↑ Bcl-2 семейство: введение. Сайт humbio.ru. Архивировано 23 августа 2011 года.
- ↑ Барышников, Шишкин, 2002, с. 54.
- ↑ Bcl-2 белки: подобие каналообразующим белкам. Сайт humbio.ru. Архивировано 23 августа 2011 года.
- ↑ Льюин и др., 2011, с. 623.
- ↑ Льюин и др., 2011, с. 626.
- ↑ Барышников, Шишкин, 2002, с. 55.
- ↑ Bcl-2 семейство: возможные механизмы действия. Сайт humbio.ru. Архивировано 23 августа 2011 года.
- ↑ FADD (Mort1) и индукция апоптоза через CD95 белок. Сайт humbio.ru. Архивировано 23 августа 2011 года.
- ↑ DR3 и DR5 рецепторы: модуляция передачи сигнала Apo2L ложными рецептор. Сайт humbio.ru. Архивировано 23 августа 2011 года.
- ↑ DcR1 (TRID, TRAIL-R3, LIT) рецептор белок. Сайт humbio.ru. Архивировано 23 августа 2011 года.
- ↑ DcR2 (TRAIL-R4 или TRUNDD) рецептор белок. Сайт humbio.ru. Архивировано 23 августа 2011 года.
- ↑ p53: функционирование. Сайт humbio.ru. Архивировано 23 августа 2011 года.
- ↑ p53 белок: Участие в апоптозе. Сайт humbio.ru. Архивировано 23 августа 2011 года.
- ↑ 1 2 Ярилин, 2003, с. 46.
- ↑ 1 2 Ярилин, 2003, с. 47.
- ↑ Ярилин, 2003, с. 48.
- ↑ Ярилин, 2003, с. 50.
- ↑ Ярилин, 2003, с. 51.
- ↑ Апоптоз и старение: общие сведения. Сайт humbio.ru. Архивировано 23 августа 2011 года.
- ↑ 1 2 Анисимов, 2008, с. 254.
- ↑ Апоптоз: возрастные изменения в неделящихся клетках. Сайт humbio.ru. Архивировано 23 августа 2011 года.
- ↑ Апоптоз: возрастные изменения в слабо пролиферирующих тканях. Сайт humbio.ru. Архивировано 23 августа 2011 года.
- ↑ Апоптоз: возрастные изменения в быстроделящихся клетках. Сайт humbio.ru. Архивировано 16 октября 2011 года.
- ↑ 1 2 3 4 5 6 7 Ярилин, 2003, с. 53.
- ↑ Фролов и др., 1999, с. 71.
- ↑ 1 2 3 Льюин и др., 2011, с. 634.
- ↑ 1 2 Ярилин, 2003, с. 52.
- ↑ 1 2 Фролов и др., 1999, с. 70.
- ↑ Барышников, Шишкин, 2002, с. 272.
- ↑ Апоптоз в эволюционном плане. Сайт humbio.ru. Архивировано 23 августа 2011 года.
- ↑ Барышников, Шишкин, 2002, с. 50.
- ↑ Шилов В. Н. (2006) Молекулярные механизмы структурного гомеостаза. Москва, издательство "Интерсигнал". 286 с., с. 156.
- ↑ 1 2 3 4 5 Льюин и др., 2011, с. 630.
- ↑ 1 2 Льюин и др., 2011, с. 629.
- ↑ Borrás et al., 2006, p. 1.
- ↑ Borrás et al., 2006, p. 1—2.
- ↑ Borrás et al., 2006, p. 3.
- ↑ Frisch S. M., Screaton R. A. Anoikis mechanisms. (англ.) // Current opinion in cell biology. — 2001. — Vol. 13, no. 5. — P. 555—562. — PMID 11544023.
- ↑ Vanden Berghe T., Linkermann A., Jouan-Lanhouet S., Walczak H., Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. (англ.) // Nature reviews. Molecular cell biology. — 2014. — Vol. 15, no. 2. — P. 135—147. — doi:10.1038/nrm3737. — PMID 24452471.
- ↑ Манских, 2007, с. 909.
Литература
- Alberts B. at al. Molecular biology of the cell. — 5th edition. — Garland science, 2008. — 1601 p. — ISBN 978-0-8153-4105.
- Banfalvi G. Apoptotic chromatin changes. — Springer science + Business media B. V., 2009. — 412 p. — ISBN 978-1-4020-9560-3.
- Borrás O. et al. Programmed cell death in plants and animals (англ.) // Biotecnología Aplicada. — 2006. — Vol. 23. — P. 1—10.
- Chen G. G., Lai P. B. S. (eds.). Apoptosis in carcinogenesis and chemotherapy. Apoptosis in cancer. — Springer, 2009. — 384 p. — ISBN 978-1-4020-9596-2.
- Lockshin R. A., Zakeri Z. (eds.). When cells die II: A comprehensive evaluation of apoptosis and programmed cell death (англ.). — John Wiley & Sons, 2004. — 572 p. — ISBN 978-0-471-21947-7.
- Vaux D. L. Apoptosis Timeline (англ.) // Cell Death and Differentiation. — 2002. — Vol. 9. — P. 349—354. — ISSN 1350-9047. — doi:10.1038/sj/cdd/4400990.
- Анисимов В. Н. Молекулярные и физиологические механизмы старения. — 2-ое, переработанное и дополненное. — СПб.: Наука, 2008. — Т. 1. — 481 с. — ISBN 978-5-02-026356-7. Архивировано 8 ноября 2011 года.
- Барышников А. Ю., Шишкин Ю. В. Иммунологические проблемы апоптоза. — М.: Эдиториал УРСС, 2002. — 320 с. — 1000 экз. — ISBN 5-8360-0328-9.
- Богадельников И. В., Вяльцева Ю. В. Поле битвы — апоптоз // Перинатология и педиатрия. — 2009. — № 3 (39). — С. 159/2.
- Ванюшин Б. Ф. Апоптоз у растений // Успехи биологической химии. — Институт физико-химической биологии им. А. Н. Белозерского, Московского государственного университета им. М. В. Ломоносова, Москва, 2001. — Т. 41. — С. 3—38.
- Гордеева А. В., Лабас Ю. А., Звягильская Р. А. Апоптоз одноклеточных организмов: механизмы и эволюция // Биохимия. — 2004. — Т. 69, вып. 10. — С. 1301—1313.
- С. Л. Кузнецов, Мушкамбаров Н. Н. Гистология, цитология и эмбриология: Учебник для медицинских вузов. — М.: ООО «Медицинское информационное агентство», 2007. — 600 с. — ISBN 5-89481-238-0.
- Льюин Б. и др. Клетки. — М.: БИНОМ. Лаборатория знаний, 2011. — 951 с. — (Лучший зарубежный учебник). — ISBN 978-5-94774-794-2.
- Манских В. Н. Пути гибели клетки и их биологическое значение // Цитология. — 2007. — Т. 49, № 11. — С. 909—915.
- Фролов В. А., Дроздова Г. А., Казанская Т. А., Билибин Д. П., Демуров Е. А. Патологическая физиология. — М.: ОАО «Издательство «Экономика», 1999. — 616 с. — ISBN 5-282-01971- X.
- Ярилин А. А. Апоптоз и его роль в целостном организме // Глаукома. — 2003. — Вып. 2. — С. 46—54.
- Сибиряк С. В., Капулер О. М., Курчатова Н. Н. и др. Апоптоз и иммунная система // Медицинский вестник Башкортостана. — 2006. — Т. 1. — № 1. — С. 127-133.
Ссылки
Иллюстрации
- Тематическая выборка иллюстраций в базе Science Photo Library (англ.). www.sciencephoto.com. Архивировано 4 августа 2012 года.
Анимация
- Институт медицинских исследований Уолтера и Элизы Холл. Fas-опосредованный апоптоз (анимированное видео) (англ.) (26 августа 2009).
- Институт медицинских исследований Уолтера и Элизы Холл. Fas-опосредованный апоптоз (анимированное видео) с русскими субтитрами (26 августа 2009).
Видео
- Тематическая выборка видеороликов в базе Science Photo Library (англ.). www.sciencephoto.com. — Для просмотра необходим QuickTime Player. Архивировано 4 августа 2012 года.
- Джошуа Гольдштейн и Дуглас Грин. Апоптоз (видео № 1) (англ.). Molecular Biology of the Cell, 5th Edition (Media DVD-ROM). Garland Science (2001).
- Джошуа Гольдштейн и Дуглас Грин. Апоптоз (видео № 2) (англ.). Molecular Biology of the Cell, 5th Edition (Media DVD-ROM). Garland Science (2001).
| В библиографических каталогах |
|---|
Типы клеточной гибели | |
|---|---|
| Непрограммируемые | |
| Программируемые | |
| Фазы |
|  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Регуляторы |
| ||||||||||
| Патогистология | |
|---|---|
| Типовые патологические процессы | |
| Лабораторная диагностика и аутопсия | |











