Атаксия телеангиэктазия
| Атаксия телеангиэктазия | |
|---|---|
| МКБ-10 | G11.3 |
| МКБ-10-КМ | G11.3 |
| МКБ-9 | 334.8 |
| МКБ-9-КМ | 334.8[1] |
| OMIM | 208900 |
| DiseasesDB | 1025 |
| MedlinePlus | 001394 |
| eMedicine | derm/691 oph/319 |
| MeSH | D001260 |
Атаксия телеангиэктазия (англ. Ataxia Telangiectasia) (АТ), известная также как синдром Луи-Бар (англ. Louis–Bar syndrome) является редким нейродегенеративным наследственным заболеванием, вызывающим тяжёлую инвалидность. Болезнь описала в 1941 г. бельгийская врач-невропатолог Дениз Луи-Бар. Атаксия приводит к плохой координации, а телеангиэктазия — слегка расширенным кровеносным сосудам; оба признака являются отличительными чертами болезни[2]. Атаксия телеангиэктазия вызывается мутациями в гене ATM[3], который отвечает за ответ клетки на двунитевые разрывы в ДНК.
При атаксии телеангиэктазии страдают различные органы и функции организма. Во-первых, при этом заболевании ухудшается работа определённых областей мозга, включая мозжечок, что вызывает трудности с движениями и координацией. Во-вторых, при АТ ослаблена иммунная система, что приводит к предрасположенности к инфекции. В-третьих, при АТ нарушена репарация ДНК, что увеличивает риск развития опухолей.
Симптомы чаще всего появляются в раннем детстве, когда дети начинают ходить. Хотя больные АТ, как правило, начинают ходить в нормальном возрасте, они раскачиваются или колеблются при ходьбе, стоя или сидя, и может показаться, что они пьяны. В конце дошкольного и младшего школьного возраста у них развиваются трудности с перемещением глаз из одного направления в другое (глазодвигательная апраксия). У них развивается невнятная или искажённая речь и глотания. Некоторые из них имеют повышенную инфекционную восприимчивость дыхательных путей (инфекции уха, синусит, бронхит и пневмония). Поскольку не все дети развиваются с одинаковой скоростью, диагностика заболевания может занять несколько лет. Большинство детей с AT имеют стабильные неврологические симптомы в течение первых 4-5 лет жизни, но ещё бóльшее число проблем у них возникает в младшем школьном возрасте.
AT вызвана мутациями в гене АТМ, который участвует в клеточном ответе на повреждение ДНК. Белок, кодируемый геном ATM, опознаёт наличие двунитевого разрыва в ДНК, после чего рекрутирует другие белки, участвующие в репарации ДНК, а также вызывает остановку клеточного цикла до тех пор, пока репарация не будет завершена[4].
Симптомы
Существует значительная вариабельность в протекании AT. Следующие симптомы являются либо общими для разных больных с АТ, либо являются важными особенностями AT:
- Атаксия (трудности с контролем движения). Проявляется рано, но ухудшается в школьном возрасте до 10-летнего возраста.
- Глазодвигательная апраксия (трудности с координацией движения головы и глаз при переключении взгляд с одного объекта на другой)
- Непроизвольные движения
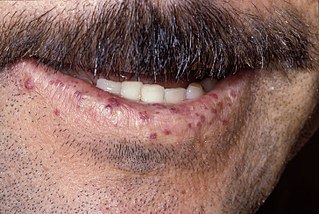
- Телеангиэктазия (расширенные кровеносные сосуды) склеры глаз, что делает её кроваво-красной. Этот признак не заметен в младенчестве и может впервые появиться в возрасте 5-8 лет. Телеангиэктазия может также появиться на открытых участках кожи.
- Инфекционные проблемы, особенно уха, придаточных пазух и лёгких.
- Увеличение числа случаев рака (в первую очередь, но не исключительно, лимфомы и лейкоза)
- Задержка начала полового развития или неполное развитие, и очень ранняя менопауза
- Замедление темпов развития (вес и/или роста)
- Слюни, особенно у маленьких детей, когда они устают или сосредотачиваются на какой-либо деятельности.
- Дизартрия (невнятная, медленная или искажённая речь)
- Диабеты в подростковом возрасте или позже
- Преждевременные изменения в волосах и коже
Многие дети первоначально неправильно диагностируются, как имеющие атаксический церебральный паралич. Диагноз AT не может быть поставлен до наступления дошкольного возраста, когда начинают появляться или ухудшаться неврологические симптомы нарушенной походки, координации рук, речи и движения глаз. Заболевание является редким, поэтому врачи не слишком хорошо знакомы с симптомами или методами постановки диагноза. Кроме того, препятствием для диагноза может быть позднее появление симптомов.
Атаксия и другие неврологические проблемы
Первые признаки AT обычно появляются в младенческом возрасте[5][6]. Дети начинают ходить в обычном возрасте, но со временем не могут избавиться от шаткости при ходьбе. Иногда у них есть проблемы колебаний из стороны в сторону, вперёд или назад, стоя или сидя. В младшем школьном возрасте ходить становится более трудно, и дети нередко используют дверные проёмы и стены для поддержки. Часто детям с AT легче двигаться быстро, чем двигаться медленно или стоять на одном месте. Начиная со второго десятилетия жизни, дети с типичными формами AT обычно вынуждены использовать инвалидное кресло для передвижения на большие расстояния. В школьные годы у детей могут возникнуть большие трудности с чтением из-за нарушения координации движения глаз. В то же время могут возникнуть другие проблемы с мелкими моторными функциями (письмо, раскрашивание и пользование ложкой при еде) и нечленораздельность речи (дизартрия). Остановить прогрессирование большинства из этих неврологических проблем удаётся после достижения возраста примерно 12 — 15 лет, хотя в любом возрасте могут начаться непроизвольные движения, которые могут ухудшаться со временем. Эти непроизвольные движения могут принимать различные формы: малые рывки рук и ног, которые выглядят как ёрзание(хорея), медленные движения со скручиванием верхней части тела (атетоз), принятие жёстких и витых поз (дистония), иногда неконтролируемые судороги (миоклонические рывки[англ.]), а также различные ритмические и неритмичные движения с попытками скоординированных действий (тремор).
Телеангиэктазия

Выделение кровеносных сосудов (телеангиэктазия) на белой склере[7] глаз, как правило, происходит в возрасте 5-8 лет, но иногда и позже, или вообще не происходит[8]. Отсутствие телеангиэктазии не исключает диагноз AT. Являясь потенциально косметической проблемой, глазная телеангиэктазия не кровоточит и не зудит, хотя она иногда ошибочно диагностируется как хронический конъюнктивит. У неё постоянная природа, не меняется со временем, с погодой или эмоциями, в отличие от других видимых повреждений кровеносных сосудов. Телеангиэктазия может появиться на солнце на открытых участках кожи, особенно на лице и ушах. Она возникает также на слизистой мочевого пузыря в качестве позднего осложнения после химиотерапии циклофосфамидом, была также замечена в глубине мозга пожилых людей с AT, а иногда возникает в печени и лёгких.
Иммунные проблемы
Около двух третей людей с AT имеют проблемы с иммунной системой[9]. Наиболее распространённые аномалии — низкие уровни одного или нескольких классов иммуноглобулинов (подклассов (IgG, IgA, IgM или IgG), отсутствие генерации антител в ответ на вакцины или инфекции и малое количество лимфоцитов (особенно Т-лимфоцитов) в крови. Некоторые люди часто страдают от инфекций верхних (простуда, пазухи и инфекции уха) и нижних (бронхит и пневмония) дыхательных путей. Все дети с AT должны пройти диагностику иммунной системы для выявления тех, у кого есть серьёзные проблемы, требующие лечения, чтобы минимизировать число и тяжесть инфекционных заболеваний. Некоторым людям с AT нужны дополнительные прививки (особенно с пневмонией и гриппом), антибиотики, чтобы обеспечить защиту от инфекций и/или инъекции иммуноглобулинов (гамма-глобулин). Потребность в этих процедурах должны быть определены с помощью специалиста в области иммунодефицита или инфекционных заболеваний.
Злокачественные опухоли
Люди с AT имеют значительный риск заболеваемости (примерно 25 % риска для жизни) раком, особенно лимфом и лейкозов, хотя могут возникнуть и другие виды рака[10]. Когда это возможно, при лечении следует избегать использования лучевой терапии и лекарств химиотерапии, которые работают по принципу, похожему на лучевую терапию (радиомиметические препараты), так как они являются токсичными, особенно для людей с AT. Специфика проблемы ведения рака достаточно сложна, так что лечение должно производиться только в академических онкологических центрах и после консультаций с врачами, прошедшим специальную подготовку к AT. К сожалению, нет способа предсказать, у каких лиц будет развиваться рак. Поскольку лейкоз и лимфомы отличаются от солидных опухолей отсутствием прогрессирования от одиночной до метастатических стадий, снижается необходимость диагностировать их в начале их появления. Надзор за лейкозом и лимфомой, таким образом, не несёт пользы, кроме рассмотрения рака как диагностический возможности, когда необъяснимо возникают возможные симптомы рака (например, стойкие опухание лимфатических желез, лихорадка).
Женщины, которые являются носителями (имеющие одну мутантную копию гена ATM), имеют примерно в два раза повышенный риск развития рака молочной железы по сравнению с населением в целом[11][12]. Это включает в себя всех детей у матерей и некоторых родственниц. Текущий консенсус состоит в том, что специальные скрининговые тесты не приносят пользы, но все женщины должны проходить рутинный надзор рака.
Кожа
АТ может вызвать признаки раннего старения, такие как преждевременное поседение волос. Она также может привести к витилиго (аутоиммунное заболевание, вызывающее потерю пигмента кожи, в результате чего возникает пятнистая кожа), и бородавкам, которые могут быть обширными и недоступные для лечения. У небольшого количества людей развиваются хронические воспалительные заболевания кожи (гранулемы)[13].
Заболевания лёгких
Хронические заболевания лёгких развивается в более чем у 25 % людей с AT[14]. Три основных типа заболевания лёгких могут развиваться: (1) рецидивирующия и хроническая синопульмонарная инфекция, (2) заболевания лёгких, вызванные неэффективностью кашля, дисфункции глотания и нарушение клиренса дыхательных путей, (3) рестриктивное интерстициальне заболевание лёгких. Это является общим для людей с AT, имеющих более одного из этих лёгких условий. Хронические заболевания лёгких могут происходить из-за повторяющихся легочных инфекций в связи с иммунодефицитом. Лица с этой проблемой имеют риск развития бронхоэктатической болезни, состояние, в котором бронхи постоянно повреждены, в результате чего — рецидивирующие инфекционные заболевания нижних дыхательных путей. Гамма-глобулин для людей с дефицитом антител и/или хроническая терапия антибиотиками может уменьшить проблемы инфекций. Другие люди с AT имеют трудности с глубоким вдохом и могут иметь неэффективный кашель, который не позволяет очистить оральные и бронхиальные секреции. Это может привести к длительным респираторным симптомам последующих общих респираторных вирусных заболеваний. Методы, которые производят очистку слизи, могут быть полезны для некоторых лиц во время респираторных заболеваний. У некоторых людей будут развиваться проблемы глотания, поскольку они стареют, а это увеличивает риск аспирационной пневмонии. Периодические повреждения лёгких, вызванных хроническими инфекциями или аспирацией, может привести к фиброзу лёгких и рубцеванию. Этот процесс может быть усилен в результате недостаточного восстановления в тканях ATM-дефицитных клеток. У небольшого количества лиц развиваются интерстициальные заболевания лёгких. Они снижают легочной резерв, затрудняют дыхание, порождают потребность в дополнительном кислороде и хронический кашель в отсутствие легочных инфекций. Они могут реагировать на системное лечение стероидами или другими препаратами для уменьшения воспаления.
Функция легочных испытаний (спирометрия), должны выполненяться, по крайней мере ежегодно у детей достаточного возраста для их выполнения, вакцины против гриппа и пневмококков следует давать при необходимости и производить лечение агрессивной синопульмонарной инфекции, чтобы ограничить развитие хронического заболевания лёгких.
Питание, глотание, и кормление
Кормление и глотание может стать трудным для людей с AT когда они становятся старше[15]. Кормление относится ко всем аспектам еды и питья, в том числе употреблению продуктов питания и жидкостей посредством рта; глотание относится к проглатыванию или тому, что происходит после того, как пища или жидкость попадает в рот. Основными задачами для кормления и глотания являются безопасные, адекватные и приятные приёмы употребления пищи.
Непроизвольные движения могут затруднять кормление и могут чрезмерно продлить время приёма пищи. Здесь может быть легче кормить пальцем, чем с использованием посуды (например, ложки или вилки). Для жидкостей, часто легче пить из закрытой ёмкости с соломинкой, чем из открытого кубка. Воспитателям, возможно, потребуется давать пищу или жидкость для самостоятельного кормления, если это возможно, иначе они должны сами накормить человека с AT. В общем, питание должно быть завершено в течение примерно 30 минут. Более длинные блюда могут вызвать стресс, мешать другой повседневной деятельности и ограничить потребление необходимых жидкостей и питательных веществ.
Если возникают проблемы глотания (дисфагия), они, как правило, присутствуют во втором десятилетии жизни. Дисфагия является общим из неврологических изменений, которые мешают координации движений рта и глотки (горла), которые необходимы для безопасного и эффективного глотания. Проблемы координации с участием рта могут затруднять жевание и увеличивать продолжительность приёма пищи. Проблемы, связанные с глотанием, могут касаться жидкостей, продуктов питания и попадания слюны в дыхательные пути (аспирация). Люди с дисфагией не могут кашлять, когда у них аспирация (тихая аспирация). Глотание и особенно глотание с тихой аспирацией может привести к проблемам с лёгкими из-за неспособности к кашлю и очистки от пищи и жидкостей дыхательных путей.
- Предупредительные признаки проблемы глотания
- Ребёнок задыхается или кашляет во время еды или питья
- Плохое увеличение веса (в возрасте от ожидаемого роста) или потеря веса в любом возрасте
- Чрезмерное слюнотечение
- Приём пищи дольше 40 — 45 минут, на регулярной основе
- Продукты и напитки, которыми ранее пользовались, теперь отказываются принимать или принимают с трудом
- Жевательная проблемы
- Увеличение частоты и продолжительности дыхания или респираторные проблемы
- Увеличение инфекционных заболеваний лёгких.
Глаз и зрение
- У большинства людей развивается телеангиэктазия (видны кровеносные сосуды) в мембране[7], покрывающей белую часть (склеру) глаза. Зрение (способность чётко видеть объекты) при этом вполне нормально[16].
- Контроль движения глаз часто нарушается, затрагивая зрительные функции, которые требуют быстрых и точных перемещений взгляда от точки к точке (например, чтение).
- Глазные смещения (косоглазие) являются общими, но могут быть излечимы.
- Возможны трудности в координации позиции глаз и кривизны хрусталика для близко расположенных объектов.
Ортопедия
У многих людей с AT развивается деформация стоп, усугубляющая трудности у них при ходьбе из-за нарушенной координации. Раннее лечение может замедлить прогрессирование этого уродства. Крепление или хирургическая коррекция иногда улучшает стабильность на лодыжке, достаточной для индивидуума, чтобы идти с поддержкой, или переносить вес на ассистента или переводить его из одной стороны на другую. Тяжелый сколиоз сравнительно редок, но, вероятно, чаще, чем у тех, кто не страдает AT. Спинное слияние бывает редко.
Патофизиология
Как потеря белка ATM создаёт мультисистемные нарушения?
Характеристики белка ATM
- 3056 аминокислот
- Серин/треонин протеинкиназа
- Член семейства киназоподобных киназ(PIKK) P13
- Размещается главным образом в ядре; небольшие количества в цитоплазме и митохондриях
- Активизируется главным образом при DSB и оксидативных стессах, но также является агентом организации хроматина и гипотонических стрессов
- Фосфорилирует и регулирует различные белковые субстраты, вовлечённые в:
- Отклик на клеточные стресы
- Контрольные точки клеточного цикла
- Репарацию ДНК
- Апоптоз
АТ была описана как синдром нестабильности генома, синдром расстройства репарации ДНК и реакции на повреждения ДНК (DDR). Ген ATM, ответственный за это мультисистемное расстройство, кодирует белок с тем же именем, который координирует клеточную реакцию на двойные разрывы нитей ДНК (DSB)[17]. Лучевая терапия, химиотерапия, которая действует как радиация (радиомиметических препараты) и некоторые биохимические процессы и метаболиты могут вызывать DSB. Когда происходят эти разрывы, ATM блокирует клетку от принятия новой ДНК (арест клеточного цикла) и рекрутирует и активирует другие белки для репарации повреждений. Таким образом, ATM позволяет клетке производить репарацию её ДНК до завершения клеточного деления. Если повреждение ДНК слишком тяжелое, ATM становится посредником в процессе запрограммированной гибели клеток (апоптоза), чтобы устранить поражённые клетки и предотвратить нестабильность генома[18].
Рак и радиочувствительность
В отсутствие белка ATM, контрольная точка регуляции клеточного цикла и запрограммированной смерти клеток в ответ на DSB оказывается дефектной. В результате возникает геномная нестабильность, которая может привести к развитию рака[25].
Облучение и радиомиметические соединения вызывают DSB, которые при отсутствии ATM не могут быть устранены соответствующим образом. Следовательно, такие агенты могут оказаться особенно цитотоксичны для AT клеток и людей с AT.
Задержка полового развития/Гонадная дисгенезия
Бесплодие часто описывается как характеристика AT. В то время как это, конечно, относится к мышиной модели AT[26], у человека она может быть более точной, чтобы охарактеризовать репродуктивную аномалию в половой атрофии или дисгенезе, характеризующимися задержкой полового развития. Поскольку запрограммированные DDR генерируются для инициации генетической рекомбинации, участвующей в производстве спермы в яйце и репродуктивных органах (процесс, известный как мейоз), мейотических дефекты и арест могут произойти при отсутствии ATM[26][27][28].
Дефекты иммунной системы и иммунно-связанный рак
ATM и иммунная система
- ATM вовлечена в:
- V(D)J рекомбинацию при производстве иммуноглобулина и T-клеток
- Переключение класса рекомбинации (изменение константного региона антител
- Пролиферации T-клеток и выживание последующей стимуляции рецептора T-клеток
- Недостаток ATM приводит к:
- Пониженному уровню иммуноглобулина(особенно подклассов IgA, IgG и IgE)
- Лимфопении (особенно проявляется в числе T-клеток
- Геномной нестабильности и транслокациям, которые могут быть результатом лимфоретикулярных нарушений
В-лимфоциты из стволовых клеток в костном мозге в зрелых лимфоцитах на периферии переставляют специальные сегменты их ДНК [V (D) J процессы рекомбинации]. Этот процесс требует от них произвести DSB, которые трудно исправить в отсутствие ATM[33][34][35][36]. В результате, у большинства людей с AT наблюдается уменьшение числа лимфоцитов и некоторое ухудшение их функции (такие как нарушения способности вырабатывать антитела в ответ на вакцины или инфекции). Кроме того, мелкие кусочки ДНК в хромосомах, участвующих в вышеупомянутых перегруппировках, имеют тенденцию к рекомбинации с другими генами (транслокации), что делает клетки склонными к развитию рака (лимфомы и лейкозы).
Прогерические изменения
Клетки людей с AT демонстрируют нестабильность генома, медленный рост и преждевременное старение в культуре, укороченные теломеры и постоянный, низкий уровень стрессового реагирования[4][37]. Эти факторы могут способствовать прогерии (признаки раннего старения), иногда наблюдаются изменения кожи и волос у людей с AT. Например, повреждения ДНК и нестабильности генома по причине дифференциации стволовых клеток меланоцитов (MSC), приводит к поседению. Таким образом, ATM может быть «контрольной точкой стволовости», защищающей от дифференциации MSC и преждевременного поседения волос[38].
Телеангиоэктазия
Причиной телеангиэктазии или расширенных кровеносных сосудов в отсутствие белка АТМ пока не известна.
Увеличение уровня альфа-фетопротеина (AFP)
Примерно 95 % людей с AT имеют повышенные уровни в сыворотке AFP после двух лет, и измеренные уровни AFP показывают медленное увеличение с течением времени[39]. Уровни AFP очень высоки у новорождённых, и, как правило, опускаются до взрослого уровня в течение от первого года до 18 месяцев. Причина, почему люди с AT имеют повышенные уровни AFP, пока не известна.
Нейродегенерация
AT является одним из нескольких расстройств репарации ДНК, которое приводит к неврологическими нарушениями или дегенерации. Возможно некоторые из самых разрушительных симптомов AT являются результатом постепенного вырождения мозжечка, характеризующегося потерей клеток Пуркинье и, в меньшей степени, зернистых клеток (находящихся исключительно в мозжечке)[5]. Причина этой потери клеток неизвестна, есть много гипотез на основе экспериментов, выполненных как в клеточной культуре, так и в мышиной модели AT. Текущие гипотезы, объясняющие нейродегенерацию, связанную с AT, включают в себя следующее:
- Дефектный отклик на повреждения ДНК в нейронах[19][40], которые могут привести к:
- Неудачному оформлению геномных повреждений нейронов во время развития[41][42]
- Неудачной транскрипции в том числе расщепления зависимых поражений комплекса топоизомеразы 1 (TOP1cc)[43][44]
- Анеуплоидии[45]
- Окислительный стресс[46][47]
- Неадекватность клеточного цикла повторного входа постмитотических (зрелых) нейронов[48]
- Синаптические/везикулярные нарушение регуляции[49] и нарушение регуляции HDAC4[50][51]
Эти гипотезы не могут быть взаимоисключающими, и более одного из этих механизмов не может лежать в основе гибели нейронов, когда отсутствует или недостаёт ATM. Кроме того, мозжечковые повреждения и потери клеток Пуркинье и зернистых клеток не объясняют все неврологические нарушения, встречаются у людей с AT. Эффекты дефицита ATM в других участках мозга вне мозжечка активно исследуются.
Воздействие радиации
Люди с AT имеют повышенную чувствительность к ионизирующей радиации (рентгеновские лучи и гамма-лучи). Таким образом, рентгеновское облучение должно быть ограничено лишь до необходимого по медицинским показаниям, поскольку ионизирующее излучение может привести к повреждению клеток таким образом, что тело не может восстановить их. Клетки могут справиться как правило, с другими формами радиации, такие как ультрафиолетовые лучи, так что нет необходимости в специальных мерах предосторожности от солнечного света.
Генетика и информация о переносчиках AT

AT вызывается мутациями в гене ATM, который был выделен в 1995 году[3]. ATM расположен на человеческой хромосоме 11 (11q22.3) и состоит из 69 экзонов, разбросанных по 150kb геномной ДНК[52].
AT наследуется аутосомно-рецессивным методом. Каждый из родителей является носителем, это означает, что они имеют одну нормальную копию гена AT (ATM) и один мутированный экземпляр. AT происходит, если ребёнок наследует мутантный ген AT от каждого родителя, так что в семье, где оба родителя являются носителями, есть 1 шанс из 4, что ребёнок, родившийся от родителей будет с нарушением. Пренатальная диагностика (и выявление переносчика) может осуществляться в семьях, если были идентифицированы ошибки (мутации) в двух генах ATM больного ребёнка. Процесс выявления этого может быть сложным и, так как это требует времени, должен быть организован до зачатия.
Выявление мутаций в гене ATM несвязанного человека (например, является ли супруг носителем AT) представляет значительные трудности. Гены часто имеют вариантные написания[53] (полиморфизмы), которые не влияют на функцию. В таком большом гене, как ATM, любой вариант написания может произойти и врачи не всегда могут предсказать, способен ли конкретный вариант вызывать болезнь. Генетическое консультирование может помочь членам семьи больного AT понять, что может или не может быть проверено, и как следует интерпретировать результаты испытаний.
Носители AT, такие как родители человека с AT, имеют одну мутантную копию гена ATM и одну нормальную копию. Они, как правило, здоровы, но есть повышенный риск рака молочной железы у женщин. Этот вывод был подтверждён различными способами и является предметом исследования в настоящее время. Рекомендуются стандартные наблюдения (в том числе ежемесячные самообследования молочных желез и обычная возрастная маммография), если не указаны дополнительные тесты, так как у человека могут быть другие факторы риска (например, семейный анамнез рака молочной железы).
Диагностика
Диагноз AT обычно предполагает комбинацию неврологических клинических признаков (атаксия, нарушение контроля движения глаз, и постуральная нестабильность) с телеангиэктазией, а иногда и с увеличением инфекционных заболеваний, и подтверждается конкретными лабораторных аномалиями (повышение уровней альфа-фетопротеина, увеличение хромосомных повреждений или гибели клеток лейкоцитов после воздействия рентгеновских лучей, отсутствие белка ATM в белых клетках крови, или мутации в каждом из генов ATM данного лица).
Разнообразие лабораторных аномалий возникают у большинства людей с AT, что позволяет делать предварительную диагностику в присутствии типичных клинических признаков. Не все аномалии наблюдаются у всех пациентов. Эти нарушения включают в себя:
- Повышение и медленное увеличение уровня альфа-фетопротеина в сыворотке крови после 2-х лет.
- Иммунодефицита с низким уровнем иммуноглобулинов (особенно подклассов IgA, IgG и IgE) с низким числом лимфоцитов в крови.
- Хромосомную нестабильность (кусков хромосом).
- Повышенная чувствительность клеток к рентгеновскому облучению (клетки умирают или производят больше разрывов и других повреждений хромосом)[54].
- Атрофия мозжечка на МРТ.
Диагноз может быть подтверждён в лаборатории путём определения отсутствия или дефицита белка ATM в культивируемых клетках крови[55][56], отсутствие или недостаток функции ATM (киназы), или мутаций в обоих копиях клеток гена ATM. Эти более специализированные тесты не всегда необходимы, но особенно полезны, если симптомы ребёнка являются нетипичными.
Дифференциальный диагноз
Есть несколько других расстройств с подобными симптомами или лабораторными признаками, которые врачи должны учитывать при диагностике AT[57]. Тремя наиболее распространёнными заболеваними, которые иногда путают с AT, являются:
- Церебральный паралич
- Атаксия Фридрейха
- Глазодвигательнная апраксия Когана.
Каждый из них можно отличить от AT при неврологическом осмотре и истории болезни.
Церебральный паралич (CP) описывает непрогрессирующее расстройство двигательной функции, вытекающее из пороков или начала повреждения мозга. СР может проявиться многими путями, представляющими различные варианты повреждения мозга; общим для всех форм появление признаков и симптомов нарушения является то, как развивается ребёнок. Тем не менее, вехи, которые были достигнуты и неврологические функции, которые были разработаны, не ухудшаются в CP, как это часто происходит у детей с AT в конце дошкольного возраста. Большинство детей с атаксией, вызванные СР не начинают ходить в нормальном возрасте, в то время как большинство детей с AT начать ходить в нормальном возрасте, хотя они часто «раскачиваются» с самого начала. Чистая атаксия является редким проявлением раннего повреждения мозга или пороков развития, однако, и возможность оккультного генетического расстройства мозга следует рассматривать и искать тех, у кого атаксия является главной манифестацией СР. Дети с атаксической CP не будет проявлять лабораторные отклонения, связанные с AT.
Глазодвигательная апраксия Когана является редким расстройством развития. Пострадавшие дети имеют трудности с передвижением глаза только на новый визуальный объект, так что они будут поворачивать голову дальше цели, чтобы «перетащить» глаз на новый интересный объект, затем поворачивают голову назад. Эта тенденция становится очевидной в конце младенчества и возраста малыша и в основном улучшается со временем. Это контрастирует с очевидными глазодвигательными трудностями детей с AT, которые не очевидны в раннем детстве, но возникают с течением времени. Глазодвигательная апраксия Когана, как правило, изолированная проблема, или может быть связаны с более широкой задержкой развития.
Атаксия Фридрейха (FA) является наиболее распространённой генетической причиной атаксии у детей. Как AT, FA является рецессивным заболеванием, появляющимся в семьях без истории нарушений. FA вызвана мутацией в гене фратаксина, чаще расширенного количества GAA повторов трёх нуклеотидных оснований, в отличие от обычных 5-33 повторов этой последовательности тринуклеотидов до более чем 65 повторов в каждой хромосоме. Чаще атаксия появляется между 10 и 15 годами, и отличается от AT отсутствием телеангиэктазии и глазодвигательной апраксии, нормальными альфа-фетопротеинами, и часто присутствием сколиоза, отсутствием сухожильных рефлексов, и аномальными особенностями на ЭКГ. Лица с FA представляются с трудом стоящими на одном месте, что значительно усиливается при закрытия глаз (знак Ромберга), что не так очевидно у лиц с AT, — хотя лица с AT могут иметь большие трудности, стоять на одном месте с открытыми глазами.
Есть другие редкие заболевания, которые можно спутать с AT, из-за аналогичных клинических признаков, сходства некоторых лабораторных признаков, или того и другого. К ним относятся:
- Атаксия глазодвигательной апраксии тип 1 (AOA1)
- Атаксия глазодвигательного апраксии тип 2 (AOA2 также известная как SCAR1)
- Атаксия телеангиэктазия, как расстройство (ATLD)
- Синдром повреждения Неймегена (NBS)
Сравнение клинических и лабораторных данных редких генетических нарушений, которые могут быть спутаны с AT (см. рисунок 3)
| AT | AOA1 | AOA2 | ATLD | NBS | |
|---|---|---|---|---|---|
| Ген | ATM | APTX | SETX | MRE11 | NBS1 |
| Радиочувствительность | DSB | SSB | Нет | DSB | DSB |
| Иммунодефицит | Да | Нет | Нет | Нет | Да |
| Нейродегенерация | Да | Да | Да | Да | Нет |
| Раннее развитие нервной системы | Обычно нормальное | Нормальное | Нормальное | Нормальное | Микроцефалия и когнитивное расстройство |
| Риск рака | Да | Нет | Нет | Неизвестно | Да |
| Альбумин | Нормальный | Низкий | Нормальный | Нормальный | Нормальный |
| AFP | Высокий | Нормальный | Высокий | Нормальный | Нормальный |
DSB — дефект репарации двойного разрыва нитей, SSB — дефект репарации одиночного разрыва.

Атаксия глазодвигательной апраксии типа 1 (AOA1) является аутосомно-рецессивным заболеванием, похожим на AT в проявлении возрастающих проблем с координацией и глазодвигательной апраксией, часто в том же возрасте, что и AT. Это вызвано мутацией в гене, кодирующем белок апратаксин. Пострадавшие люди отличаются от лиц с AT ранним появлением периферической невропатии, ранним демонстрированием трудностей с началом сдвигов взгляда и отсутствием глазной телеангиэктазии, но лабораторные особенности имеют ключевое значение в их различиях. Лица с AOA1 имеют нормальную AFP, нормальную иммунную функцию и после 10-15 лет имеют низкие уровни альбумина в сыворотке. Генетический анализ гена апратаксин может подтвердить диагноз. У них отсутствует повышенный риск для рака.
Атаксия глазодвигательной апраксия тип 2 (AOA2) является аутосомно-рецессивным заболеванием, также похожим на AT в проявлении возрастающих проблем с координацией и периферической невропатией, но глазодвигательная апраксия присутствует только у половины пострадавших лиц. Глазная телеангиэктазия у них не развиваются. Лабораторные аномалии AOA2, как и AT, в отличие от AOA1, заключаются в повышенном уровне AFP в сыворотке, но, отличие от AOA1 и AT — в нормальных маркерах иммунной функции. Генетическое тестирование гена сенатаксин (SETX) может подтвердить диагноз. Повышенного риска для рака нет.
Атаксия телеангиэктазия-как расстройство (ATLD) является чрезвычайно редким заболеванием, вызванным мутацией в гене hMre11, которая может быть рассмотрена в дифференциальной диагностике AT. Пациенты с ATLD очень похожи пациентов AT демонстрацией прогрессирующей мозжечковой атаксии, повышенной чувствительностью к ионизирующей радиации и нестабильностью генома. Те немногие люди с ATLD, которые хорошо описаны отличаются от людей с AT отсутствием телеангиэктазии, нормальным уровнем иммуноглобулинов в позднем возрасте и в более медленном прогрессировании симптомов. Из-за своей редкости, ещё не известно, несёт ли ATLD повышенный риск развития рака. Поскольку мутации Mre11, что серьёзно ухудшают белок MRE11, несовместимы с жизнью, люди с ATLD обладают частичной функцией белка Mre11, и, следовательно, скорее всего, все они имеют свои собственные уровни тяжести заболевания.
Синдром повреждения Неймегена (NBS) является редким генетическим заболеванием, аналогичным хромосомной нестабильности, что наблюдается у людей с AT, но возникающие проблемы совсем другие. Дети с NBS обладают значительнойй микроцефалией, выделюяшимся внешний видом лица, низкорослостью и умеренным когнитивным ухудшением, но не испытывают неврологического ухудшения в течение долгого времени. Как и лица с AT, у детей с NBS повышенная чувствительность к радиации, склонность к лимфоме и лейкозу и некоторым лабораторным показателям нарушения иммунной функции, но отсутствует глазная телеангиэктазия или повышенный уровень AFP.
Интересно, что белки, экспрессируемые генами hMre11 (дефектный в ATLD) и Nbs1 (дефектный в NBS) существуют в клетке в виде комплекса, вместе с третьим белком, экспрессированным геном hRad50. Этот комплекс, известный как комплекс MRN, играет важную роль в репарации повреждений и сигнализации и требуется для рекрутирования ATM на сайты двухцепочечных разрывов ДНК. Mre11 и Nbs1 также ориентированы на фосфорилирование киназы ATM. Таким образом, сходство трёх заболеваний можно частично объяснить тем, что белковые продукты трёх мутированных генов в этих расстройствах имеют в клетке общие пути взаимодействия.
Дифференциацию этих расстройств часто можно произвести посредством учёта клинических особенностей и проведения отдельных лабораторных тестов. В тех случаях, когда различие неясно, клинические лаборатории могут выявить генетические аномалии ATM, апратаксина и сенатаксина и специализированные центры могут определить аномалии белков потенциально ответственных генов, таких как ATM, MRE11, нибрин, TDP1, апратаксин и сенатаксин, а также других белков, важных для функции ATM, таких как ATR, ДНК-ПК, и RAD50.
Ведение
Атаксия и другие неврологические проблемы
Не существует, как известно, лечения, чтобы замедлить или остановить прогрессирование неврологических проблем. Лечение AT лишь симптоматическое и поддерживающее. Физическая, профессиональная и речевая терапия и физические упражнения могут помочь сохранить функцию, но не смогут замедлить ход нейродегенерации. Лечебная гимнастика не должна быть использована до точки усталости и не должна вмешиваться в повседневную жизнь. Некоторые антипаркинсоновские и противоэпилептические лекарственные средства могут быть полезными в управлении симптомами, но должны быть предписаны в результате консультации с неврологом.
Иммунные проблемы
Все лица с AT должны проходить по крайней мере одну комплексную иммунологическую проверку, которая измеряет количество и тип лимфоцитов в крови (Т-лимфоцитов и В-лимфоцитов), уровни сывороточных иммуноглобулинов (IgG, IgA, IgM и антител) и отклики антител на Т-зависимые (например, столбняк, грипп Hemophilus b) и Т-независимые (23-валентная пневмококковая полисахаридная) вакцины. По большей части, какая картина иммунодефицита видна у AT пациента в начале жизни (в возрасте до пяти), она будет такой же видна в течение всей жизни этого лица. Таким образом, испытания не обязаны повторяться, если у этого лица не увеличиваются инфекционные проблемы. Проблемы с иммунитетом иногда могут быть преодолены с помощью иммунизации. Вакцины против общих бактериальных респираторных патогенов, таких как грипп Hemophilus, пневмококки и вирус гриппа («flu»), коммерчески доступны и часто помогают повысить иммунные реакции, даже у лиц с низким уровнем иммуноглобулина. Если вакцины не работают и у пациента продолжаются инфекционные проблемы, то гамма-глобулиновая терапия (IV или подкожная инфузия антител, полученных от здоровых людей) может быть полезной. У некоторого количества людей с AT развивается аномалии, при которых один или более типов иммуноглобулина повышен далеко за пределами нормального диапазона. В некоторых случаях, уровни иммуноглобулинов могут быть увеличены настолько, что кровь становится густой и утрачивает необходимую текучесть. Терапия для этой проблемы должны быть адаптированы к конкретным найденным нарушениям и их тяжести.
Если восприимчивость инфекционных заболеваний отдельного пациента увеличивается, важно пересмотреть направление терапии в случае ухудщения иммунных функций. Если инфекционный проблемы происходят в лёгких, необходимо изучить возможность неблагополучного глотания с аспирацией в лёгкие (смотри выше подразделы Симптомы: Заболевания лёгких и Симптомы: Питание глотание и кормление ).
Большинство людей с AT имеют низкие показатели лимфоцитов в крови. Эта проблема, как представляется, относительно стабильна с возрастом, лишь малое число людей имеют постепенно уменьшающееся с возрастом число лимфоцитов. В общей популяции очень низкое число лимфоцитов связано с повышенным риском инфицирования. У таких людей развиваются осложнения к живых вирусным вакцинам (против кори, эпидемического паротита, краснухи и ветрянки), хроническим или тяжелым вирусным инфекциям, дрожжевым инфекциям кожи и вагины, и оппортунистическим инфекциям (таким, как пневмоцистная пневмония). Хотя число лимфоцитов часто занижено у людей с AT, они редко имеют проблемы с оппортунистическими инфекциями. (Одно исключение из этого правила является то, что проблемы с хроническими или рецидивирующими бородавками являются общими). Функции и количество Т-лимфоцитов должны быть пересмотрены, если человека с AT обрабатывают кортикостероидными препаратами, такими как преднизолон, дольше, чем несколько недель или обрабатывают раковой химиотерапией. Если количество лимфоцитов является низким у людей, принимающих эти виды препаратов, рекомендуется использование профилактических антибиотиков для предотвращения воздействия оппортунистических инфекций.
Если тесты показывают существенные нарушения иммунной системы, специалист в области иммунодефицитных или инфекционных заболеваний будет иметь возможность обсудить различные варианты лечения. Отсутствие отклика иммуноглобулина или антител на вакцину можно лечить с помощью замены гамма-глобулина вливаниями, или можно вести профилактическими антибиотиками и свести к минимуму воздействие инфекции. Если функция антител в норме, должны быть проведены все рутинные виды иммунизации детей в том числе живыми вирусными вакцинами (против кори, эпидемического паротита, краснухи и ветряной оспы). Кроме того, несколько «специальных» вакцин (то есть, лицензированных не для обычных здоровых детей и молодых взрослых) следует использовать, чтобы уменьшить риск развития у пациента с AT легочных инфекционных заболеваний. Пациент и все члены семьи должны получать вакцины против гриппа каждую осень. Люди с AT, которые моложе двух лет должны получать три (3) дозы пневмококковой конъюгированной вакцины (Prevnar[англ.]), даваемой с двухмесячным интервалом. Люди старше двух лет, которым ранее не были сделаны прививки с Prevnar должны получить две (2) дозы Prevnar. По крайней мере, через 6 месяцев после последней прививки Prevnar должна быть введена ребёнку, по крайней мере двухлетнему, 23-валентная пневмококковая вакцина. Иммунизация 23-валентной пневмококковой вакциной необходимо повторить примерно через каждые пять лет после первой дозы.
У людей с AT, которые имеют низкий уровень IgA, должно быть выполнено дальнейшее тестирование, чтобы определить, низкий уровень IgA или полностью отсутствует. Если отсутствуют, есть несколько повышенный риск реакции на переливание. Браслеты «Медицинская тревога» не являются необходимыми, но семья и лечащий врач должны знать, что, если выбранная хирургия требует переливания эритроцитов, клетки должны быть вымыты, чтобы уменьшить риск аллергической реакции.
Люди с AT также имеют повышенный риск развития аутоиммунных или хронических воспалительных заболеваний. Этот риск, вероятно, побочный эффект их иммунодефицита, а не прямое воздействие недостатка белка ATM. Наиболее распространённые примеры таких иммунных нарушений при AT включают в себя такие, как тромбоцитопения (ITP), несколько форм артрита, и витилиго.
Заболевания лёгких
Периодические инфекции пазухи и лёгких могут привести к развитию хронического заболевания лёгких[14]. Такие инфекции следует лечить соответствующими антибиотиками по предотвращению и ограничению травм лёгких. Назначение антибиотиков следует рассматривать, когда дети и взрослые имеют длительные респираторные симптомы (более 7 дней), даже после предположения, что была вирусная инфекция. Чтобы предотвратить заболевания дыхательных путей от общих респираторных патогенов, следует при необходимости делать ежегодные прививки грипповой и пневмококковой вакцины. Лечение антибиотиками также следует предусмотреть для детей с хроническими кашлем, являющимся влажным, тех, кому не помогают энергичные методы легочной очистки и детей со слизисто-гнойными выделениями из пазухи или груди. Влажный кашель также может быть связан с хронической аспирацией, которая должна быть исключена посредством соответствующих диагностических исследований, однако аспирация и респираторные инфекции не обязательно исключают друг друга. У детей и взрослых с бронхоэктазами, следует применять хроническую антибактериальную терапию, чтобы замедлить прогрессирование хронического заболевания лёгких.
Культивирование пазух может быть необходимо для направления терапии антибиотиками. Это может быть сделано с помощью специалиста уха, горла и носа (ЛОР). Кроме того, диагностическая бронхоскопия может быть необходима у людей, которые имеют текущие пневмонии, особенно у тех, кто не реагирует или не полностью реагирует на курс антибиотиков.
Клиренс бронхиального секрета имеет важное значение для хорошего здоровья лёгких и может помочь ограничить травмы от острых и хронических легочных инфекций. Дети и взрослые с повышенным бронхиальным секретом могут извлечь выгоду из обычной терапии груди с помощью ручного метода, устройства капелла или жилета физиотерапии грудной клетки. Физиотерапия груди может помочь поднять слизистый секрет от нижнего бронхиального дерева, однако необходим адекватной кашель для удаления выделений. У людей с пониженным объёмом лёгких и слабым кашлем, может быть полезно использование устройства инсуффлятор-экссуфлятор (помощник кашля) в качестве поддерживающей терапии или при острых респираторных заболеваних, чтобы помочь удалить бронхиальные секреции из верхних дыхательных путей. Однако, прежде нужно сделать оценку специалистом пульмонологии, чтобы правильно оценить пригодность пациента.
Дети и взрослые с хроническим сухим кашлем, повышенной работой органов дыхания (быстрый темп дыхания, одышка в покое или в деятельности) и отсутствием инфекционного процесса, для объяснения респираторных симптомов должны быть оценены на наличие интерстициальной болезни лёгких или другого внутрилегочного процесса. Оценку пульмонолога и КТ грудной клетки следует произвести у пациентов с симптомами интерстициального заболевания лёгких или другого неинфекционного легочного процесса. Людям с диагнозом интерстициального заболевания лёгких могут помочь системные стероиды.
Кормление, глотание и питание
Устная беседа может способствовать обучению лиц с AT, как пить, жевать и глотать более безопасно. Правильность лечения для глотания должна быть определена после оценки экспертом в области патологии речевого языка. Диетологи могут помочь лечить проблемы питания, рекомендуя диетические изменения, в том числе в плане питания высококалорийными продуктами или пищевыми добавками.
Рекомендуется использование трубки кормления (гастростомии) в любом из следующих случаев[58]:
- Ребёнок или человек любого возраста не может съесть достаточно, чтобы расти или поддерживать вес;
- Аспирация пищевых масс;
- Приём пищи напряжённый или слишком долгий, мешает другим видам деятельности.
Трубки кормления могут уменьшить риск аспирации, позволяя лицам избегать жидкостей или продуктов, которые трудно проглотить и предоставить необходимое количество калорий без стресса и длительного времени употребления еды. Трубки гастростомии не мешают людям есть через рот. После того, как трубка находится на месте, общая цель должна заключаться в поддержании веса на 10-25 перцентилей.
Образование и социализация
Большинство детей с AT испытывают трудности в школе из-за задержки во времени реакции на визуальные, вербальные или другие сигналы, невнятной и тихой речи (дизартрия), нарушения управления глаз (глазодвигательная апраксия) и нарушение точного управления движением. Несмотря на эти проблемы, дети с AT часто посещают школу, если могут быть сделаны соответствующие жилые помещения с учётом их инвалидности. Решение о необходимости специальных классов образования или дополнительной помощи в обычных классах во многом зависит от местных ресурсов. Решения о надлежащих учебных заведениях должны быть пересматриваемы так часто, как того требуют обстоятельства. Несмотря на многие неврологических нарушения, большинство людей с AT очень социальны и, таким образом, им будет польза от устойчивых отношений со сверстниками в школе. Некоторые люди способны функционировать достаточно хорошо, несмотря на недостатки физиологии и некоторые из них окончили колледжи.
Многие из проблем, которые требуют особого внимания, часто больше связаны с возможностью управления, чем интеллектуальными нарушениями. Проблемы с контролем движения глаз для людей с AT затрудняет чтение, задерживая восприятие смысла и нюансов текста. Задержки в инициации речи и отсутствия мимики приводят к представлению, что они не знают ответы на вопросы. Увеличение имеющегося времени, чтобы ответить, часто вознаграждается реальным выполнением задачи. Важно признать, что интеллектуальная инвалидность не является регулярной частью клинической картины AT, хотя успеваемость в школе может быть неоптимальной из-за многих трудностей с чтением, письмом и речью. Дети с AT очень часто сознают их наличие, а также стремятся не отставать от нормальных своих сверстников и преподавателей. Жизнь с атаксией может быть утомительной. Увеличиваются усилия, необходимые для поддержания внешнего вида и требуется повышенная энергия, расходуемая в аномальных дополнительных движениях, всё это способствуют физической и умственной усталости. Как следствие, для некоторых сокращение учебного дня приносит реальную пользу.
Общие рекомендации :
- Всем детям с AT необходимо уделять особое внимание к барьерам, которые они испытывают в школе. В Соединённых Штатах, это принимает форму официального ИПО (Индивидуальной программы обучения).
- Дети с AT, как правило, отлично решают свои проблемы. Их участие в том, как лучше выполнять задачи, следует поощрять.
- Патологи языковой речи могут способствовать развитию коммуникативных навыков, которые позволяют людям с AT получать сообщения (с помощью ключевых слов или полных предложений) и научить стратегии снижения фрустраций по поводу увеличения времени для ответа на вопросы (например, подняв руку и другие виды запросов о необходимости увеличения времени для ответов). Редко бывают полезны традиционные речевые методы лечения, которые сосредоточены на производстве определённых звуков и укрепления мышц и губ языка.
- Использование классных помощников вполне целесообразно, особенно, чтобы помочь со скрайбированием, передвижением внутри школы, приёмом пищи и пользованием туалетом. Влияние помощника на взаимоотношения со сверстниками должно тщательно контролироваться.
- Физическая терапия полезна для поддержания силы и общего здоровья сердечно-сосудистой системы. Верховая терапия и упражнения в бассейне часто хорошо переносится и доставляет удовольствие для людей с AT. Тем не менее, никакое количество практики не сможет замедлить мозжечковую дегенерацию или улучшить неврологические функции. Нагрузок до точки истощения следует избегать.
- Нормальный слух сохраняется в течение всей жизни. Аудиокниги могут быть полезным дополнением к традиционным школьным материалам.
- Раннее использование компьютеров (дошкольных) с программным обеспечением, завершающим слово, следует поощрять.
- Практика координации (например на бревне или рукописные письменные упражнения) не приносят пользы.
- Трудотерапия является полезной для управления навыками повседневной жизни.
- Следует предоставлять время отдыха, укороченный день, снижение плотности расписания занятий, уменьшение домашних заданий, модификацию тестов по мере необходимости.
- Как и всем детям, детям с AT необходимо иметь цели, чтобы испытывать прогресс удовлетворения.
- Социальные взаимодействия со сверстниками важны, и должны быть приняты во внимание для размещения в классе. Долгосрочные взаимоотношения со сверстниками могут продолжаться часть жизни; установление этих связей в школьные годы может быть полезным.
Клиники и поддержка
США, Великобритания, Австралия, Израиль, Нидерланды, Германия, Польша, Норвегия и Япония имеют специализированные клиники для пациентов с АТ. Эти клиники — дом многопрофильных медицинских бригад, в том числе неврологов, пульмонологов, иммунологов и врачей, способных справиться со многими проявлениями этого заболевания.
Эпидемиология
Люди всех рас и этнических групп страдают в равной степени. Заболеваемость в мире, по оценкам, от 1 в 40000 до 1 в 100000 человек[4][59].
Прогнозы
Средняя продолжительность жизни людей с AT сильно варьирует. Средняя примерно 25 лет, но продолжает улучшаться с достижениями в области оказания помощи. Две наиболее распространённые причины смерти — хронические заболевания лёгких (около одной трети случаев) и рака (около одной трети случаев).
Направления исследований
Открытая фаза II клинических испытаний, изучающая использование красных кровяных клеток (эритроцитов), загруженных с дексаметазоном фосфата натрия обнаружила, что это улучшает лечение симптомов и, кажется, хорошо переносится[60]. Эта процедура использует уникальную систему транспортировки лекарств с помощью собственных красных кровяных клеток пациента, в качестве средства доставки препарата[61]. Учитывая другие иммунологические дефициты у лиц с AT, требуется дальнейшая оценка терапевтического потенциала стероидов, особенно в отношении продолжительности какой-либо пользы и его долгосрочной безопасности.
Примечания
- ↑ Monarch Disease Ontology release 2018-06-29 — 2018-06-29 — 2018.
- ↑ Boder, E. Ataxia-telangiectasia: an overview. (неопр.) // Kroc Foundation series. — 1985. — Т. 19. — С. 1—63. — PMID 2415689.
- ↑ 1 2 3 Savitsky, K; Bar-Shira, A., Gilad, S., Rotman, G., Ziv, Y., Vanagaite, L., Tagle, D.A., Smith, S., Uziel, T., Sfez, S., Ashkenazi, M., Pecker, I., Frydman, M., Harnik, R., Patanjali, S.R., Simmons, A., Clines, G.A., Sartiel, A., Gatti, R.A., Chessa, L., Sanal, O., Lavin, M.F., Jaspers, N.G., Taylor, A.M., Arlett, C.F., Miki, T., Weissman, S.M., Lovett, M., Collins, F.S., Shiloh, Y. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. (англ.) // Science : journal. — 1995. — 23 June (vol. 268, no. 5218). — P. 1749—1753. — doi:10.1126/science.7792600. — PMID 7792600.
- ↑ 1 2 3 Shiloh, Y; Kastan, M.B. ATM: genome stability, neuronal development, and cancer cross paths. (англ.) // Advances in cancer research : journal. — 2001. — Vol. 83. — P. 209—254. — doi:10.1016/s0065-230x(01)83007-4. — PMID 11665719.
- ↑ 1 2 Crawford, T.O. Ataxia telangiectasia. (неопр.) // Seminars in pediatric neurology. — 1998. — December (т. 5, № 4). — С. 287—294. — doi:10.1016/s1071-9091(98)80007-7. — PMID 9874856.
- ↑ Crawford, TO; Mandir, A.S., Lefton-Greif, M.A., Goodman, S.N., Goodman, B.K., Sengul, H., Lederman, H.M. Quantitative neurologic assessment of ataxia-telangiectasia. (англ.) // Neurology[англ.]. — Wolters Kluwer[англ.], 2000. — 11 April (vol. 54, no. 7). — P. 1505—1509. — doi:10.1212/wnl.54.7.1505. — PMID 10751267.
- ↑ 1 2 По-видимому, здесь имеется в виду конъюнктива, в ней находятся кровеносные сосуды. (прим. переводчика)
- ↑ Cabana, MD; Crawford, TO; Winkelstein, JA; Christensen, JR; Lederman, H.M. Consequences of the delayed diagnosis of ataxia-telangiectasia. (англ.) // Pediatrics[англ.] : journal. — American Academy of Pediatrics[англ.], 1998. — July (vol. 102, no. 1 Pt 1). — P. 98—100. — doi:10.1542/peds.102.1.98. — PMID 9651420.
- ↑ Nowak-Wegrzyn, A; Crawford, TO; Winkelstein, JA; Carson, KA; Lederman, H.M. Immunodeficiency and infections in ataxia-telangiectasia (англ.) // The Journal of Pediatrics[англ.] : journal. — 2004. — April (vol. 144, no. 4). — P. 505—511. — doi:10.1016/j.jpeds.2003.12.046. — PMID 15069401.
- ↑ Reiman, A; Srinivasan, V., Barone, G., Last, J.I., Wootton, L.L., Davies, E.G., Verhagen, M.M., Willemsen, M.A., Weemaes, C.M., Byrd, P.J., Izatt, L., Easton, D.F., Thompson, D.J., Taylor, A.M. Lymphoid tumours and breast cancer in ataxia telangiectasia; substantial protective effect of residual ATM kinase activity against childhood tumours. (англ.) // British Journal of Cancer[англ.] : journal. — 2011. — 9 August (vol. 105, no. 4). — P. 586—591. — doi:10.1038/bjc.2011.266. — PMID 21792198. — PMC 3170966.
- ↑ Thompson, D; Duedal, S; Kirner, J; McGuffog, L; Last, J; Reiman, A; Byrd, P; Taylor, M; Easton, D.F. Cancer risks and mortality in heterozygous ATM mutation carriers. (англ.) // Journal of the National Cancer Institute[англ.] : journal. — 2005. — 1 June (vol. 97, no. 11). — P. 813—822. — doi:10.1093/jnci/dji141. — PMID 15928302.
- ↑ Renwick, A; Thompson, D., Seal, S., Kelly, P., Chagtai, T., Ahmed, M., North, B., Jayatilake, H., Barfoot, R., Spanova, K., McGuffog, L., Evans, D.G., Eccles, D., Breast Cancer Susceptibility Collaboration, (UK), Easton, D.F., Stratton, M.R., Rahman, N. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. (англ.) // Nature Genetics : journal. — 2006. — August (vol. 38, no. 8). — P. 873—875. — doi:10.1038/ng1837. — PMID 16832357.
- ↑ Paller, AS; Massey, R.B., Curtis, M.A., Pelachyk, J.M., Dombrowski, H.C., Leickly, F.E., Swift, M. Cutaneous granulomatous lesions in patients with ataxia-telangiectasia. (англ.) // The Journal of Pediatrics[англ.] : journal. — 1991. — December (vol. 119, no. 6). — P. 917—922. — doi:10.1016/s0022-3476(05)83043-4. — PMID 1960607.
- ↑ 1 2 McGrath-Morrow, SA; Gower, W.A., Rothblum-Oviatt, C., Brody, A.S., Langston, C., Fan, L.L., Lefton-Greif, M.A., Crawford, T.O., Troche, M., Sandlund, J.T., Auwaerter, P.G., Easley, B., Loughlin, G.M., Carroll, J.L., Lederman, H.M. Evaluation and management of pulmonary disease in ataxia-telangiectasia. (англ.) // Pediatric pulmonology : journal. — 2010. — September (vol. 45, no. 9). — P. 847—859. — doi:10.1002/ppul.21277. — PMID 20583220.
- ↑ Lefton-Greif, MA; Crawford, T.O., Winkelstein, J.A., Loughlin, G.M., Koerner, C.B., Zahurak, M., Lederman, H.M. Oropharyngeal dysphagia and aspiration in patients with ataxia-telangiectasia. (англ.) // The Journal of Pediatrics[англ.] : journal. — 2000. — February (vol. 136, no. 2). — P. 225—231. — doi:10.1016/s0022-3476(00)70106-5. — PMID 10657830.
- ↑ Farr, AK; Shalev, B; Crawford, TO; Lederman, HM; Winkelstein, JA; Repka, M.X. Ocular manifestations of ataxia-telangiectasia. (англ.) // American Journal of Ophthalmology[англ.]. — 2002. — December (vol. 134, no. 6). — P. 891—896. — doi:10.1016/s0002-9394(02)01796-8. — PMID 12470759.
- ↑ 1 2 Derheimer, FA; Kastan, M.B. Multiple roles of ATM in monitoring and maintaining DNA integrity. (англ.) // FEBS Letters[англ.] : journal. — 2010. — 10 September (vol. 584, no. 17). — P. 3675—3681. — doi:10.1016/j.febslet.2010.05.031. — PMID 20580718. — PMC 2950315.
- ↑ 1 2 Kurz, EU; Lees-Miller, S.P. DNA damage-induced activation of ATM and ATM-dependent signaling pathways. (англ.) // DNA repair : journal. — Vol. 3, no. 8—9. — P. 889—900. — doi:10.1016/j.dnarep.2004.03.029. — PMID 15279774.
- ↑ 1 2 Dar, I; Biton, S; Shiloh, Y; Barzilai, A. Analysis of the ataxia telangiectasia mutated-mediated DNA damage response in murine cerebellar neurons. (англ.) // The Journal of neuroscience: the official journal of the Society for Neuroscience : journal. — 2006. — 19 July (vol. 26, no. 29). — P. 7767—7774. — doi:10.1523/JNEUROSCI.2055-06.2006. — PMID 16855104.
- ↑ Gorodetsky, E; Calkins, S; Ahn, J; Brooks, P.J. ATM, the Mre11/Rad50/Nbs1 complex, and topoisomerase I are concentrated in the nucleus of Purkinje neurons in the juvenile human brain. (англ.) // DNA repair : journal. — 2007. — November (vol. 6, no. 11). — P. 1698—1707. — doi:10.1016/j.dnarep.2007.06.011. — PMID 17706468. — PMC 2797317.
- ↑ Valentin-Vega, YA; Maclean, K.H., Tait-Mulder, J., Milasta, S., Steeves, M., Dorsey, F.C., Cleveland, J.L., Green, D.R., Kastan, M.B. Mitochondrial dysfunction in ataxia-telangiectasia. (англ.) // Blood?!. — American Society of Hematology[англ.], 2012. — 9 February (vol. 119, no. 6). — P. 1490—1500. — doi:10.1182/blood-2011-08-373639. — PMID 22144182. — PMC 3286212.
- ↑ Guo, Z; Kozlov, S; Lavin, MF; Person, MD; Paull, T.T. ATM activation by oxidative stress. (англ.) // Science. — 2010. — 22 October (vol. 330, no. 6003). — P. 517—521. — doi:10.1126/science.1192912. — PMID 20966255.
- ↑ Bakkenist, CJ; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. (англ.) // Nature : journal. — 2003. — 30 January (vol. 421, no. 6922). — P. 499—506. — doi:10.1038/nature01368. — PMID 12556884.
- ↑ Kanu, N; Behrens, A. ATMINistrating ATM signalling: regulation of ATM by ATMIN. (англ.) // Cell cycle (Georgetown, Tex.) : journal. — 2008. — 15 November (vol. 7, no. 22). — P. 3483—3486. — doi:10.4161/cc.7.22.7044. — PMID 19001856.
- ↑ Shiloh, Y. ATM and related protein kinases: safeguarding genome integrity. (англ.) // Nature Reviews Cancer. — 2003. — March (vol. 3, no. 3). — P. 155—168. — doi:10.1038/nrc1011. — PMID 12612651.
- ↑ 1 2 Barlow, C; Hirotsune, S., Paylor, R., Liyanage, M., Eckhaus, M., Collins, F., Shiloh, Y., Crawley, J.N., Ried, T., Tagle, D., Wynshaw-Boris, A. Atm-deficient mice: a paradigm of ataxia telangiectasia. (англ.) // Cell. — Cell Press, 1996. — 12 July (vol. 86, no. 1). — P. 159—171. — doi:10.1016/S0092-8674(00)80086-0. — PMID 8689683.
- ↑ Plug, AW; Peters, A.H., Xu, Y., Keegan, K.S., Hoekstra, M.F., Baltimore, D., de Boer, P., Ashley, T. ATM and RPA in meiotic chromosome synapsis and recombination. (англ.) // Nature Genetics : journal. — 1997. — December (vol. 17, no. 4). — P. 457—461. — doi:10.1038/ng1297-457. — PMID 9398850.
- ↑ Barchi, M; Roig, I., Di Giacomo, M., de Rooij, D.G., Keeney, S., Jasin, M. ATM promotes the obligate XY crossover and both crossover control and chromosome axis integrity on autosomes. (англ.) // PLOS Genetics[англ.] : journal. — 2008. — 23 May (vol. 4, no. 5). — P. e1000076. — doi:10.1371/journal.pgen.1000076. — PMID 18497861. — PMC 2374915.

- ↑ Lumsden, JM; McCarty, T., Petiniot, L.K., Shen, R., Barlow, C., Wynn, T.A., Morse H.C., 3rd, Gearhart, P.J., Wynshaw-Boris, A., Max, E.E., Hodes, R.J. Immunoglobulin class switch recombination is impaired in Atm-deficient mice. (англ.) // Journal of Experimental Medicine[англ.] : journal. — Rockefeller University Press[англ.], 2004. — 1 November (vol. 200, no. 9). — P. 1111—1121. — doi:10.1084/jem.20041074. — PMID 15504820. — PMC 2211853.
- ↑ Franco, S; Alt, FW; Manis, J.P. Pathways that suppress programmed DNA breaks from progressing to chromosomal breaks and translocations. (англ.) // DNA repair : journal. — 2006. — 8 September (vol. 5, no. 9—10). — P. 1030—1041. — doi:10.1016/j.dnarep.2006.05.024. — PMID 16934538.
- ↑ Callén, E; Jankovic, M., Wong, N., Zha, S., Chen, H.T., Difilippantonio, S., Di Virgilio, M., Heidkamp, G., Alt, F.W., Nussenzweig, A., Nussenzweig, M. Essential role for DNA-PKcs in DNA double-strand break repair and apoptosis in ATM-deficient lymphocytes. (англ.) // Molecular Cell[англ.] : journal. — 2009. — 15 May (vol. 34, no. 3). — P. 285—297. — doi:10.1016/j.molcel.2009.04.025. — PMID 19450527. — PMC 2709792.
- ↑ Bagley, J; Singh, G; Iacomini, J. Regulation of oxidative stress responses by ataxia-telangiectasia mutated is required for T cell proliferation. (англ.) // Journal of Immunology (Baltimore, Md.: 1950) : journal. — 2007. — 15 April (vol. 178, no. 8). — P. 4757—4763. — doi:10.4049/jimmunol.178.8.4757. — PMID 17404255.
- ↑ Bredemeyer, AL; Sharma, G.G., Huang, C.Y., Helmink, B.A., Walker, L.M., Khor, K.C., Nuskey, B., Sullivan, K.E., Pandita, T.K., Bassing, C.H., Sleckman, B.P. ATM stabilizes DNA double-strand-break complexes during V(D)J recombination. (англ.) // Nature : journal. — 2006. — 27 July (vol. 442, no. 7101). — P. 466—470. — doi:10.1038/nature04866. — PMID 16799570.
- ↑ Bredemeyer, AL; Huang, CY; Walker, LM; Bassing, CH; Sleckman, B.P. Aberrant V(D)J recombination in ataxia telangiectasia mutated-deficient lymphocytes is dependent on nonhomologous DNA end joining. (англ.) // Journal of Immunology (Baltimore, Md.: 1950) : journal. — 2008. — 15 August (vol. 181, no. 4). — P. 2620—2625. — doi:10.4049/jimmunol.181.4.2620. — PMID 18684952.
- ↑ Bredemeyer, AL; Helmink, B.A., Innes, C.L., Calderon, B., McGinnis, L.M., Mahowald, G.K., Gapud, E.J., Walker, L.M., Collins, J.B., Weaver, B.K., Mandik-Nayak, L., Schreiber, R.D., Allen, P.M., May, M.J., Paules, R.S., Bassing, C.H., Sleckman, B.P. DNA double-strand breaks activate a multi-functional genetic program in developing lymphocytes. (англ.) // Nature : journal. — 2008. — 11 December (vol. 456, no. 7223). — P. 819—823. — doi:10.1038/nature07392. — PMID 18849970. — PMC 2605662.
- ↑ Callén, E; Jankovic, M., Difilippantonio, S., Daniel, J.A., Chen, H.T., Celeste, A., Pellegrini, M., McBride, K., Wangsa, D., Bredemeyer, A.L., Sleckman, B.P., Ried, T., Nussenzweig, M., Nussenzweig, A. ATM prevents the persistence and propagation of chromosome breaks in lymphocytes. (англ.) // Cell : journal. — Cell Press, 2007. — 13 July (vol. 130, no. 1). — P. 63—75. — doi:10.1016/j.cell.2007.06.016. — PMID 17599403.
- ↑ Shiloh, Y; Tabor, E; Becker, Y. Colony-forming ability of ataxia-telangiectasia skin fibroblasts is an indicator of their early senescence and increased demand for growth factors. (англ.) // Experimental cell research : journal. — 1982. — July (vol. 140, no. 1). — P. 191—199. — doi:10.1016/0014-4827(82)90169-0. — PMID 6213420.
- ↑ Inomata, K; Aoto, T., Binh, N.T., Okamoto, N., Tanimura, S., Wakayama, T., Iseki, S., Hara, E., Masunaga, T., Shimizu, H., Nishimura, E.K. Genotoxic stress abrogates renewal of melanocyte stem cells by triggering their differentiation. (англ.) // Cell : journal. — Cell Press, 2009. — 12 June (vol. 137, no. 6). — P. 1088—1099. — doi:10.1016/j.cell.2009.03.037. — PMID 19524511.
- ↑ Stray-Pedersen, A; Borresen-Dale, A.L., Paus, E., Lindman, C.R., Burgers, T., Abrahamsen, T.G. Alpha fetoprotein is increasing with age in ataxia-telangiectasia. (англ.) // European journal of paediatric neurology: EJPN: official journal of the European Paediatric Neurology Society : journal. — 2007. — November (vol. 11, no. 6). — P. 375—380. — doi:10.1016/j.ejpn.2007.04.001. — PMID 17540590.
- ↑ Biton, S; Dar, I; Mittelman, L; Pereg, Y; Barzilai, A; Shiloh, Y. Nuclear ataxia-telangiectasia mutated (ATM) mediates the cellular response to DNA double strand breaks in human neuron-like cells. (англ.) // Journal of Biological Chemistry : journal. — 2006. — 23 June (vol. 281, no. 25). — P. 17482—17491. — doi:10.1074/jbc.M601895200. — PMID 16627474.
- ↑ Herzog, KH; Chong, MJ; Kapsetaki, M; Morgan, JI; McKinnon, P.J. Requirement for Atm in ionizing radiation-induced cell death in the developing central nervous system. (англ.) // Science : journal. — 1998. — 15 May (vol. 280, no. 5366). — P. 1089—1091. — doi:10.1126/science.280.5366.1089. — PMID 9582124.
- ↑ Lee, Y; Chong, MJ; McKinnon, P.J. Ataxia telangiectasia mutated-dependent apoptosis after genotoxic stress in the developing nervous system is determined by cellular differentiation status. (англ.) // The Journal of neuroscience: the official journal of the Society for Neuroscience : journal. — 2001. — 1 September (vol. 21, no. 17). — P. 6687—6693. — PMID 11517258.
- ↑ Sordet, O; Redon, C.E., Guirouilh-Barbat, J., Smith, S., Solier, S., Douarre, C., Conti, C., Nakamura, A.J., Das, B.B., Nicolas, E., Kohn, K.W., Bonner, W.M., Pommier, Y. Ataxia telangiectasia mutated activation by transcription- and topoisomerase I-induced DNA double-strand breaks. (англ.) // EMBO Reports[англ.] : journal. — 2009. — August (vol. 10, no. 8). — P. 887—893. — doi:10.1038/embor.2009.97. — PMID 19557000. — PMC 2726680.
- ↑ Das, BB; Antony, S; Gupta, S; Dexheimer, TS; Redon, CE; Garfield, S; Shiloh, Y; Pommier, Y. Optimal function of the DNA repair enzyme TDP1 requires its phosphorylation by ATM and/or DNA-PK. (англ.) // The EMBO Journal : journal. — 2009. — 2 December (vol. 28, no. 23). — P. 3667—3680. — doi:10.1038/emboj.2009.302. — PMID 19851285. — PMC 2790489.
- ↑ Iourov, IY; Vorsanova, SG; Liehr, T; Kolotii, AD; Yurov, Y.B. Increased chromosome instability dramatically disrupts neural genome integrity and mediates cerebellar degeneration in the ataxia-telangiectasia brain. (англ.) // Human Molecular Genetics[англ.] : journal. — Oxford University Press, 2009. — 15 July (vol. 18, no. 14). — P. 2656—2669. — doi:10.1093/hmg/ddp207. — PMID 19414482.
- ↑ Barzilai, A; Biton, S; Shiloh, Y. The role of the DNA damage response in neuronal development, organization and maintenance. (англ.) // DNA repair : journal. — 2008. — 1 July (vol. 7, no. 7). — P. 1010—1027. — doi:10.1016/j.dnarep.2008.03.005. — PMID 18458000.
- ↑ Biton, S; Barzilai, A; Shiloh, Y. The neurological phenotype of ataxia-telangiectasia: solving a persistent puzzle. (англ.) // DNA repair : journal. — 2008. — 1 July (vol. 7, no. 7). — P. 1028—1038. — doi:10.1016/j.dnarep.2008.03.006. — PMID 18456574.
- ↑ Yang, Y; Herrup, K. Loss of neuronal cell cycle control in ataxia-telangiectasia: a unified disease mechanism. (неопр.) // The Journal of neuroscience: the official journal of the Society for Neuroscience. — 2005. — 9 March (т. 25, № 10). — С. 2522—2529. — doi:10.1523/JNEUROSCI.4946-04.2005. — PMID 15758161.
- ↑ Li, J; Han, YR; Plummer, MR; Herrup, K. Cytoplasmic ATM in neurons modulates synaptic function. (англ.) // Current biology: CB : journal. — 2009. — 29 December (vol. 19, no. 24). — P. 2091—2096. — doi:10.1016/j.cub.2009.10.039. — PMID 19962314. — PMC 2805770.
- ↑ Nuclear accumulation of HDAC4 in ATM deficiency promotes neurodegeneration in ataxia telangiectasia. (англ.) // Nature Medicine : journal. — 2012. — May (vol. 18, no. 5). — P. 783—790. — doi:10.1038/nm.2709. — PMID 22466704.
- ↑ ATM and the epigenetics of the neuronal genome. (неопр.) // Mech Ageing Dev. — 2013. — October (т. 134, № 10). — С. 434—439. — doi:10.1016/j.mad.2013.05.005. — PMID 23707635.
- ↑ Gatti, R.A.; Berkel I., Boder E., Braedt G., Charmley P., Concannon P., Ersoy F., Foroud T., Jaspers N.G., Lange K., et al. Localization of an ataxia-telangiectasia gene to chromosome 11q22-23 (англ.) // Nature. — 1988. — Vol. 336, no. 6199. — P. 577—580. — doi:10.1038/336577a0. — PMID 3200306.
- ↑ В тексте variant spellings — здесь по-видимому имеются в виду «варианты транскрипции»; имеет место результат неудачного выбора английских синонимов вместо термина, по всей видимости (прим. переводчика)
- ↑ Sun, X; Becker-Catania, S.G., Chun, H.H., Hwang, M.J., Huo, Y., Wang, Z., Mitui, M., Sanal, O., Chessa, L., Crandall, B., Gatti, R.A. Early diagnosis of ataxia-telangiectasia using radiosensitivity testing. (англ.) // The Journal of Pediatrics[англ.] : journal. — 2002. — June (vol. 140, no. 6). — P. 724—731. — doi:10.1067/mpd.2002.123879. — PMID 12072877.
- ↑ Chun, HH; Sun, X; Nahas, SA; Teraoka, S; Lai, CH; Concannon, P; Gatti, R.A. Improved diagnostic testing for ataxia-telangiectasia by immunoblotting of nuclear lysates for ATM protein expression. (англ.) // Molecular Genetics and Metabolism[англ.] : journal. — 2003. — December (vol. 80, no. 4). — P. 437—443. — doi:10.1016/j.ymgme.2003.09.008. — PMID 14654357.
- ↑ Taylor, AM; Byrd, P.J. Molecular pathology of ataxia telangiectasia. (англ.) // Journal of Clinical Pathology[англ.]. — 2005. — October (vol. 58, no. 10). — P. 1009—1015. — doi:10.1136/jcp.2005.026062. — PMID 16189143. — PMC 1770730.
- ↑ Anheim, M; Tranchant, C; Koenig, M. The autosomal recessive cerebellar ataxias. (англ.) // The New England Journal of Medicine. — 2012. — 16 February (vol. 366, no. 7). — P. 636—646. — doi:10.1056/NEJMra1006610. — PMID 22335741.
- ↑ Lefton-Greif, MA; Crawford, TO; McGrath-Morrow, S; Carson, KA; Lederman, H.M. Safety and caregiver satisfaction with gastrostomy in patients with Ataxia Telangiectasia. (англ.) // Orphanet Journal of Rare Diseases[англ.] : journal. — 2011. — 15 May (vol. 6). — P. 23. — doi:10.1186/1750-1172-6-23. — PMID 21569628. — PMC 3116459.
- ↑ Swift, M; Morrell, D; Cromartie, E; Chamberlin, AR; Skolnick, MH; Bishop, D.T. The incidence and gene frequency of ataxia-telangiectasia in the United States. (англ.) // American Journal of Human Genetics[англ.] : journal. — 1986. — November (vol. 39, no. 5). — P. 573—583. — PMID 3788973. — PMC 1684065.
- ↑ Chessa, L; Leuzzi, V; Plebani, A; Soresina, A; Micheli, R; D'Agnano, D; Venturi, T; Molinaro, A; Fazzi, E; Marini, M; Ferremi Leali, P; Quinti, I; Cavaliere, FM; Girelli, G; Pietrogrande, MC; Finocchi, A; Tabolli, S; Abeni, D; Magnani, M. Intra-erythrocyte infusion of dexamethasone reduces neurological symptoms in ataxia teleangiectasia patients: results of a phase 2 trial. (англ.) // Orphanet Journal of Rare Diseases[англ.] : journal. — 2014. — 9 January (vol. 9, no. 1). — P. 5. — doi:10.1186/1750-1172-9-5. — PMID 24405665.
- ↑ Yousefpour, P; Chilkoti, A. Co-opting biology to deliver drugs. (англ.) // Biotechnology and Bioengineering[англ.]. — 2014. — September (vol. 111, no. 9). — P. 1699—1716. — doi:10.1002/bit.25307. — PMID 24916780.
Ссылки
- The A-T Children’s Project
- Wobbly Feet Foundation, Inc.
- The UK AT Society
- Action for A-T Charity
- Asociacion Espanola Familia Ataxia-Telangiectasia (AEFAT)
- BrAshA-T, Australia
- Projeto AT/Brasil
- Association Pour la Recherche sur l’A-T (APRAT)
- AT Europe
- AT Info, Germany
- Un Vero Sorriso Onlus (A-True Smile Association)
- Gli Amici di Valentina (Friends of Valentina)
- Team A-T, Japan
- Stichting A-T, Netherlands
- Twan Foundation, Netherlands
- Razem Zdazymy (Together We are in Time)
- FEAT An independent documentary to raise awareness for A-T
- About A-T from the NINDS
- Orphanet for A-T
- GeneReviews for Ataxia-Telangiectasia
- Replication-Independent Double-Strand Breaks (DSBs) Discusses importance of the ATM kinase
- Cancer.Net: Ataxia-Telangiectasia