
Боле́знь По́мпе — редкое, наследственное заболевание с аутосомно-рецессивным механизмом, наследования,, связанное с повреждением мышечных, и нервных клеток по всему организму. Клиническая картина данной патологии, обусловлена накоплением гликогена, в лизосомах, вызванным недостаточностью лизосомного фермента — кислой α-1,4-глюкозидазы. Различают быстро прогрессирующую (классическую) и медленно прогрессирующую формы болезни Помпе. Несмотря на широкий спектр клинических проявлений, гликогеноза II типа, в основе всех форм болезни лежит, дефицит одного фермента, кодируемого геном GAA.
Муколипидозы — собирательное название группы наследственных заболеваний, относящихся к лизосомным болезням накопления, связанных с дефицитом того или иного фермента. Гетерогенная группа болезней, объединяющая проявления недостаточности одного из ферментов лизосом, результатом которого является определённое сочетание накопления внутри клеток организма мукополисахаридов, гликопротеинов, олигосахаридов и гликолипидов. Изначально данная группа генных болезней, клиническая картина которых связана с нарушением нормального катаболизма различных субстратов внутри клеток, названа по аналогии с другими наследственными болезнями накопления. Открытие биохимических процессов, дефект которых приводил к развитию того или иного типа муколипидоза, внёс корректировку в классификацию. Изначально первые четыре типа были помечены как муколипидозы. Однако, теперь муколипидоз I типа (сиалидоз) классифицируется как гликопротеиноз, а муколипидоз IV типа (сиалолипидоз) — как ганглиозидоз.
Боле́знь Бильшо́вского — Янско́го — поздняя инфантильная (детская) форма восковидного липофусциноза нейронов, которая развивается на фоне дефицита лизосомного фермента трипептидил-пептидазы-1. Относится к группе лизосомных болезней накопления.

Метахромати́ческая лейкодистрофи́я — редкое наследственное заболевание из группы лизосомных болезней накопления с аутосомно-рецессивным механизмом наследования нарушения обмена веществ. Данная нозологическая единица из разряда лейкодистрофий относится к сфинголипидозам. Характеризуется недостаточностью арилсульфатазы А (цереброзидсульфатазы) — фермента лизосом, участвующего в метаболизме сфинголипидов, что вызывает накопление цереброзида сульфата.
Синдро́м Шейе — тяжёлое наследственное заболевание из группы мукополисахаридозов, относящихся к лизосомным болезням накопления. Характеризуется недостаточностью альфа-L-идуронидазы — фермента лизосом, участвующего в катаболизме кислых мукополисахаридов, которые составляют основу межклеточного вещества соединительной ткани. Заболевание редкое, проявляется в детском возрасте.
Синдро́м Гу́рлер — Шейе́ — наследственное заболевание из группы мукополисахаридозов, относящихся к лизосомным болезням накопления. Характеризуется недостаточностью альфа-L-идуронидазы — фермента лизосом, участвующего в катаболизме кислых мукополисахаридов, которые составляют основу межклеточного вещества соединительной ткани. Заболевание редкое, проявляется в детском возрасте.
Синдром Марото — Лами — редкая наследственная болезнь, одна из форм мукополисахаридоза из группы лизосомных болезней накопления, биохимически связанная с дефицитом фермента лизосом N-Ацетилгексозамин-4-сульфатсульфатазы.

GM2-ганглиозидо́з — тяжёлое наследственное заболевание, развивающееся в результате дефицита или недостаточной активности фермента гексозаминидазы и накопления в клетках ганглиозидов. Относится к лизосомным болезням накопления и имеет три варианта, связанные с мутациями в разных генах, которые оказывают вляние на активность общей гексозаминидазы. Два варианта заболевания больше известны под индивидуальными именами, полученными в честь авторов, впервые описавших их клиническую картину: болезнь Тея — Сакса, болезнь Сандхоффа. Третий вариант этой болезни носит название GM2 ганглиозидоз, вариант АБ. Болезнь Тея — Сакса вызвана мутацией в гене HEXA, кодирующий альфа-субъединицу гексоаминидазы А. Болезнь Сандхоффа вызвана мутацией в гене HEXB, кодирующий бета-субъединицу гексоаминидаз А и Б. GM2 ганглиозидоз, АБ вариант связан с нарушением в гене GM2, кодирующий белок-активатор GM2A.
GM2 ганглиозидо́з, вариа́нт АБ (англ. GM2-gangliosidosis, AB variant) — редкое аутосомно-рецессивное нарушение обмена веществ, связанное с мутацией гена GM2A. Характеризуется нормальной активностью β-гексозаминидаз А и Б и обусловлено недостаточностью активатора (белкового кофактора), необходимого для реализации ферментной активности в отношении субстрата. Заболевание клинически проявляется прогрессирующим разрушением нервных клеток головного и спинного мозга.
GM1-ганглиозидо́зы — редкие наследственные заболевания из группы лизосомных болезней накопления. Развитие клинической картины обусловлено дефектом или недостатком β-галактозидазы, который ведёт к нарушению метаболизма и накоплению субстратов (ганглиозида GM1, гликопротеинов и кератансульфата) главным образом в нервных клетках центральной и периферической нервной системы.

Синдром Сандхоффа — наследственное заболевание, вариант 0 GM2-ганглиозидоза из группы лизосомных болезней накопления — нарушение липидного обмена, вызванное наследственным дефицитом (недостаточностью) ферментов бета-гексозаминидазы А и Б.
Синдро́м Сла́я — редкое наследственное заболевание из группы мукополисахаридозов, относящееся к лизосомным болезням накопления, характеризуется дефицитом фермента лизосом β-глюкуронидазы. В свою очередь, дефицит β-глюкуронидазы ведёт к накоплению определённых сложных углеводов (мукополисахаридов) во многих тканях и органах тела человека.
Псевдополидистрофи́я Гу́рлер — наследственное заболевание из группы муколипидозов, относящееся к лизосомным болезням накопления с аутосомно-рецессивным механизмом наследования нарушения обмена веществ. Клинически представляет собой заболевание с фенотипическими признаками мукополисахаридоза, напоминает более лёгкий вариант I-клеточной болезни. Данный симптомокомплекс получил название псевдо-Гурлер в связи с клинической схожестью симптомов с одним из представителей мукополисахаридоза — синдромом Гурлер.
Недоста́точность ки́слой фосфата́зы — редкое наследственное заболевание из группы лизосомных болезней накопления. Клиническая симптоматика дефицита кислой фосфатазы развивается в раннем возрасте. Характеризуется гепатомегалией и задержкой психического развития.

Маннозидо́зы — редкие наследственные заболевания из группы лизосомных болезней накопления с аутосомно-рецессивным типом наследования.
Боле́знь Ку́фса — редкое наследственное нейродегенеративное заболевание с летальным исходом из группы лизосомных болезней накопления, которое развивается на фоне дефицита фермента лизосом и наследуется по аутосомно-рецессивному типу.
Пикнодизосто́з — редкое наследственное заболевание из группы лизосомных болезней накопления, патология костной ткани, связанная с мутацией гена, кодирующего фермент катепсин K.
Аспартилглюкозаминури́я — редкое аутосомно-рецессивное наследственное заболевание из группы лизосомных болезней накопления, связанное с нарушением метаболизма олигосахаридов в результате снижения активности фермента лизосом аспартилглюкозаминамидазы.

Фукозидо́з — редкое наследственное заболевание из группы лизосомных болезней накопления, связанное с недостаточностью гидролаз, расщепляющих полисахаридные связи. В результате внутри клеток накапливаются два класса макромолекул: гликолипиды и гликопротеины. Клиническая картина связана с мутацией, приводящей к повреждению или дефициту фермента лизосом альфа-L-фукозидазы и характеризуется разнообразными соматическими проявлениями и нарушением функции нервной системы.
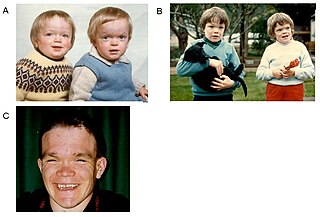
Альфа-маннозидо́з — редкое аутосомно-рецессивное наследственное заболевание из группы лизосомных болезней накопления, связанное с нарушением метаболизма олигосахаридов в результате снижения активности фермента лизосом альфа-маннозидазы. Также заболевание наносит ущерб в животноводстве.
![Ангиокератома туловища — клинический признак, ассоциированный с β-маннозидозом[1]](https://upload.wikimedia.org/wikipedia/commons/thumb/8/85/Angiokreatoma.jpg/274px-Angiokreatoma.jpg)
