
Боле́знь Паркинсо́на — медленно прогрессирующее хроническое нейродегенеративное неврологическое заболевание, характерное для лиц старшей возрастной группы. Относится к дегенеративным заболеваниям экстрапирамидной моторной системы. Вызвано прогрессирующим разрушением и гибелью нейронов, вырабатывающих нейромедиатор дофамин, — прежде всего в чёрной субстанции, а также и в других отделах центральной нервной системы. Недостаточная выработка дофамина ведёт к тормозному влиянию базальных ганглиев на кору головного мозга. Ведущими симптомами являются:
- мышечная ригидность;
- гипокинезия;
- тремор;
- постуральная неустойчивость.
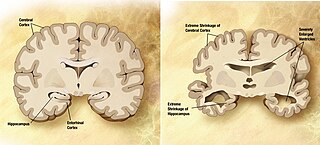
Боле́знь Альцгеймера — наиболее распространённая форма деменции, нейродегенеративное заболевание, впервые описанное в 1907 году немецким психиатром Алоисом Альцгеймером (1864—1915). Как правило, она обнаруживается у людей старше 65 лет, но существует и ранняя болезнь Альцгеймера — редкая форма заболевания. Общемировая заболеваемость на 2006 год оценивалась в 26,6 млн человек, а к 2050 году число больных может вырасти вчетверо.

Болезнь Гентингтона — аутосомно-доминантное генетическое заболевание нервной системы, характеризующееся постепенным началом обычно в возрасте 30—50 лет и сочетанием прогрессирующего хореического гиперкинеза и психических расстройств. Заболевание вызывается умножением кодона CAG в гене HTT. Этот ген кодирует 350-kDa белок хантингтин с неизвестной функцией. В гене дикого типа у разных людей присутствует разное количество CAG-повторов, однако, когда число повторов превышает 36, развивается болезнь. Нейроморфологическая картина характеризуется атрофией стриатумa, а на поздней стадии также атрофией коры головного мозга.
Рассе́янный склеро́з (РС), или множественный склероз, — хроническое аутоиммунное заболевание, при котором поражается миелиновая оболочка проводников головного и спинного мозга. Хотя в разговорной речи «склерозом» часто называют нарушение памяти в пожилом возрасте, название «рассеянный склероз» не имеет отношения ни к старческому «склерозу», ни к рассеянности внимания. «Склероз» в данном случае означает «рубец», а «рассеянный» означает «множественный», поскольку отличительная особенность болезни при патологоанатомическом исследовании — наличие рассеянных по всей центральной нервной системе без определённой локализации очагов склероза — замены нормальной нервной ткани на соединительную. Рассеянный склероз впервые описал в 1868 году Жан-Мартен Шарко.

Мигрень — первичная форма головной боли, симптомами которой являются периодические приступы головной боли средней и высокой интенсивности. Головная боль, как правило, локализована в одной половине головы, имеет пульсирующий характер и длится от нескольких часов до 2—3 дней.. Сопутствующие симптомы включают тошноту, рвоту, гиперчувствительность к свету, звукам и запахам. Иногда боль усиливается при физической активности. Примерно у трети пациентов наблюдается аура, как правило, в виде кратковременного нарушения зрения, сигнализирующего о приближающемся приступе головной боли. Иногда аура может возникать перед слабой головной болью или независимо от неё.
Болезнь Вильсона — Коновалова — врождённое или приобретенное нарушение метаболизма меди, приводящее к тяжелейшим поражениям центральной нервной системы и внутренних органов. Диагностируется у 5-10 % больных циррозом печени дошкольного и школьного возраста. Заболевание передаётся по аутосомно-рецессивному типу. Ген ATP7B, мутации которого вызывают заболевание, расположен на 13-й хромосоме.
Ацерулоплазминемия — редкое заболевание, вызванное мутациями гена, кодирующего синтез церулоплазмина. Данный белок, основная функция которого заключается в транспорте меди, также участвует в метаболизме железа, и мутации, нарушающие эту функцию церулоплазмина, приводят к токсичным отложениям железа в мозге, печени, поджелудочной железе.
Болезнь Краббе — редкое наследственное заболевание, при котором поражается миелиновая оболочка нервных волокон. Наследуется аутосомно-рецессивно. Болезнь названа в честь датского невролога Краббе, который описал её в 1916 году.

Сэмюель Вильсон — английский невропатолог.

Болезнь Баттена — редкое смертельное нейродегенеративное рецессивно наследуемое заболевание, начинающееся в детстве, наиболее частое из группы восковидных липофусцинозов нейронов (ВЛН), относящихся к лизосомным болезням накопления. Хотя обычно это название употребляется в отношении заболевания с детства, некоторые учёные называют так все ВЛН. Исторически восковидные липофусцинозы нейронов делятся на детские, позднедетские, юношеские и взрослые. В ходе заболевания в клетках нервной системы скапливаются жировые вещества, что приводит к потере речи, нарушениям зрения, слабоумию, эпилепсии.
Нейродегенеративные заболевания — группа в основном медленно прогрессирующих, наследственных или приобретённых заболеваний нервной системы. Общим для этих заболеваний является прогрессирующая гибель нервных клеток (нейродегенерация), ведущая к различным неврологическим симптомам — прежде всего, к деменции и нарушению движений. Заболевания могут наступить в различном возрасте, протекают диффузно или генерализированно, гистологически определяется специфический тип изменений.

Демиелинизирующие заболевания — это любые заболевания нервной системы при которых повреждается миелиновая оболочка нейронов. Это повреждение нарушает проведение сигналов по пострадавшим нервам. В свою очередь, снижение проводящей способности приводит к нарушениям чувствительности, движений, когнитивных функций, и других функций в зависимости от того, какие затронуты нервы.

Сфинголипидо́зы группа лизосомных болезней накопления, связанных с нарушением метаболизма сфинголипидов, относится к классу болезней накопления липидов (липидозов). Основными представителями этой группы являются болезнь Ниманна — Пика, болезнь Фабри, болезнь Краббе, болезнь Гоше, болезнь Тея — Сакса и метахроматическая лейкодистрофия. Они, как правило, наследуется по аутосомно-рецессивному типу, однако в частности, болезнь Фабри — редкое генетически детерминированное заболевание с Х-сцепленным рецессивным типом наследования.
Наследственная оптическая нейропатия LHON Лебера, или атрофия зрительного нерва Лебера , является наследственной митохондриальной дегенерацией ганглионарных клеток (РСК) сетчатки и их аксонов, что приводит к острой или почти острой потере центрального зрения; это влияет преимущественно на молодых мужчин. Тем не менее, LHON передается только по материнской линии, прежде всего, из-за мутаций в митохондриальном геноме, и только яйцеклетка способствует митохондрии в зародыше. LHON, как правило, связана с одной из трех патогенных митохондриальных ДНК (мтДНК) точечных мутаций. Эти мутации действуют на нуклеотиды и репозиционируют 11778 G в А, 3460 G в А и 14484 T в C, соответственно, в ND4, ND1 и Nd6 субъединицах генов в комплексе I окислительного фосфорилирования цепочек в митохондриях. Мужчины не могут передать болезнь своему потомству.

Метахромати́ческая лейкодистрофи́я — редкое наследственное заболевание из группы лизосомных болезней накопления с аутосомно-рецессивным механизмом наследования нарушения обмена веществ. Данная нозологическая единица из разряда лейкодистрофий относится к сфинголипидозам. Характеризуется недостаточностью арилсульфатазы А (цереброзидсульфатазы) — фермента лизосом, участвующего в метаболизме сфинголипидов, что вызывает накопление цереброзида сульфата.
Друзы оптического диска (ODD) или друзы глазного нерва (ONHD) —глобулы из мукопротеинов и мукополисахаридов, которые постепенно кальцифицируют диск зрительного нерва. Они могут стать остатками транспортной системы аксонов вырождающихся ганглиозных клеток сетчатки. ODD также приписывались такие проблемы как врождённые поднятые или аномальные диски, псевдопапиллоэдема, псевдоневрит, утопление друз диска, и гиалиновые тельца в оптическом диске. Они могут быть связаны с потерей зрения различной степени и иногда приводить к слепоте, или, чаще, иметь бессимптомное течение.
Синдром Коккейна , также называемый синдром Нил-Дингуолл — редкое аутосомно-рецессивное, нейродегенеративное расстройство, характеризующееся недостатком роста, нарушением развития нервной системы, аномальной чувствительностью к солнечному свету (фотосенсибилизация), заболеваниями глаз и преждевременным старением. Нездоровый вид и неврологические расстройства являются критериями для диагностики, а светочувствительность, нарушения слуха и ненормальные глаза — другие весьма общие черты. Возможны проблемы любого или всех внутренних органов. Это связано с группой расстройств, называемых лейкодистрофия. В основе расстройства лежит дефект механизма репарации ДНК. Интересно, в отличие от других дефектов репарации ДНК, пациенты с CS не предрасположены к раку или инфекции. Синдром Коккейна редок, но это разрушительная болезнь, которая, как правило, приводит к смерти в первом или втором десятилетии жизни. Мутация специфических генов в синдроме Коккейна известна, но широко распространенные эффекты и его отношения с репарацией ДНК еще не очень хорошо исследованы.

Дегенеративно-дистрофическое заболевание диска (ДДЗД) описывает дисфункцию межпозвоночного диска человека. Несмотря на терминологию, ДДЗД не является ни объединённым заболеванием, ни прогрессирующим дегенеративным расстройством. В противоположность этому, дегенерация диска — это часто встречающийся эффект ежедневного стресса и небольших повреждений, которые приводят к постепенному обезвоживанию фиброзного кольца или жесткой внешней оболочки диска, и к её ослаблению. При обезвоживании дисков также происходит их ослабление и инициируются дегенеративные процессы. Суммарно, эти процессы под действием нагрузки на позвоночник приводят к сдавливанию корешков cпинного мозга, и сопровождаются сильными болями и слабостью.

Болезнь Шарко — Мари — Тута (ШМТ), или наследственная моторно-сенсорная нейропатия (НМСН) — наследственная периферическая нейропатия с хроническим прогрессирующим течением. При этой болезни больные страдают от слабости и атрофии мышц дистальных отделов конечностей, деформации стоп и кистей, у них наблюдается снижение сухожильных рефлексов, изменение походки, потеря чувствительности в конечностях. В основе клинических проявлений болезни лежит поражение двигательных и чувствительных периферических нервных волокон. Болезнь Шарко — Мари — Тута диагностируется с приблизительной частотой 1 на 2500 человек. Первое проявление болезни чаще всего происходит в подростковом возрасте или в раннем взрослом возрасте. Тяжесть симптомов сильно различается даже среди членов одной семьи с этим заболеванием. Болезнь Шарко — Мари — Тута нередко ведёт к ограничению трудоспособности и к инвалидизации, при этом большинство пациентов имеют нормальную продолжительность жизни. Болезнь Шарко — Мари — Тута является генетически крайне неоднородным заболеванием, симптомы этой болезни могут быть вызваны мутациями в более чем двух десятках генов, хотя большая часть заболеваний вызвана мутациями в генах PMP22, MPZ, GJB1 и MFN2. Наследование болезни чаще всего аутосомно-доминантное, однако может быть аутосомно-рецессивным и Х-сцепленным.
Лейкодистрофии - группа тяжелых, наследуемых преимущественно по аутосомно-рецессивному типу, заболеваний характеризующихся преимущественно поражением белого вещества головного и спинного мозга.