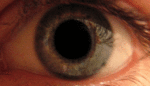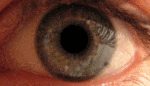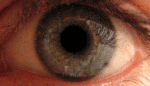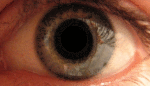
Глаз — сенсорный орган животных, обладающий способностью воспринимать электромагнитное излучение в видимом диапазоне длин волн и обеспечивающий функцию зрения. У человека через глаз поступает около 90 % информации из окружающего мира.
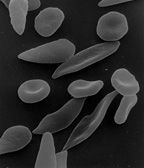
Серповидноклеточная анемия — наследственная гемоглобинопатия, связанная с таким нарушением строения белка гемоглобина, при котором он приобретает особое кристаллическое строение. Форма гемоглобина больных — так называемый гемоглобин S. Эритроциты, несущие гемоглобин S вместо нормального гемоглобина А, имеют характерную серпообразную форму, за что эта форма гемоглобинопатии и получила название серповидноклеточной анемии.

Альбинизм — наследственное заболевание, полное или почти полное отсутствие пигмента меланина или хлорофилла. Проявляется отсутствием нормальной вида окраски кожи, волос, шерсти, радужной и пигментной оболочек глаз, зелёных частей растений.

Отслойка сетчатки — отделение слоя палочек и колбочек, то есть нейроэпителия, от пигментного эпителия сетчатки, обусловленное скоплением жидкости между ними. В здоровом глазу они тесно соприкасаются.

Зрение человека — способность человека воспринимать информацию путём преобразования энергии электромагнитного излучения светового диапазона, осуществляемая зрительной системой.

Синдро́м Альстрёма — генетическая патология человека, относящаяся к группе цилиопатий. Характеризуется пигментной дегенерацией сетчатки, ожирением, прогрессирующей нейросенсорной глухотой, дилятационной кардиомиопатией, сахарным диабетом и нефропатией. Впервые описан в 1959 году шведским психиатром Карлом-Генри Альстрёмом.

Болезнь Гиппеля — Линдау — факоматоз, при котором гемангиобластомы мозжечка сочетаются с ангиомами спинного мозга, множественными врождёнными кистами поджелудочной железы и почек. У четверти больных развивается карцинома почки, часто первично-множественная. Симптомы заболевания становятся очевидными во 2-м десятилетии жизни — одним из первых обнаруживается кровоизлияние в глазное яблоко или в заднюю черепную ямку с признаками внутричерепной гипертензии или мозжечковыми расстройствами. У большинства пациентов в цереброспинальной жидкости обнаруживают повышение содержания белка, у половины детей с опухолями мозжечка — увеличение числа клеток.

Ретинопати́я недоно́шенных — тяжёлое заболевание глаз, развивающееся преимущественно у глубоконедоношенных детей, сопровождающееся изменениями в сетчатке и стекловидном теле. Появление симптома «белого зрачка» или лейкокории требует проведения тщательного диагностического поиска.

Никтало́пи́я или гемерало́пи́я, иначе — Куриная слепота — расстройство, при котором затрудняется или пропадает способность видеть в сумерках. Является симптомом ряда глазных болезней; может быть врождённым или приобретенным. В последнем случае может являться одним из симптомов заболеваний сетчатой оболочки или зрительного нерва или стать результатом неправильного питания.

Глаз человека — парный сенсорный орган зрительной системы, обладающий способностью воспринимать электромагнитное излучение в световом диапазоне длин волн и обеспечивающий функцию зрения. Глаза расположены в передней части головы и вместе с веками, ресницами и бровями, являются важной частью лица. Область лица вокруг глаз активно участвует в мимике. Максимальный оптимум дневной чувствительности человеческого глаза приходится на максимум непрерывного спектра солнечного излучения, расположенный в «зелёной» области 550 (556) нм. При переходе от дневного освещения к сумеречному происходит перемещение максимума световой чувствительности по направлению к коротковолновой части спектра, и предметы красного цвета кажутся чёрными, синего (василёк) — очень светлыми.

Ночно́е зре́ние — механизм восприятия света зрительной системой человека, действующий в условиях относительно низкой освещённости. Осуществляется с помощью палочек при яркости фона менее 0,01 кд/м2, что соответствует ночным условиям освещения. Колбочки в этих условиях не функционируют, поскольку для их возбуждения не хватает интенсивности света. Синонимы: скотопическое и палочковое зрение.

Хориоретинит — воспаление сосудистой оболочки и сетчатки глаза. Это форма заднего увеита. Если воспаляется только сосудистая оболочка и не воспаляется сетчатка, состояние называется хориоидит . Офтальмологическая цель в лечении этих болезней, потенциально опасных слепотой, — это устранение воспаления и сведение к минимуму потенциального риска терапии для пациента.
GM2 ганглиозидо́з, вариа́нт АБ (англ. GM2-gangliosidosis, AB variant) — редкое аутосомно-рецессивное нарушение обмена веществ, связанное с мутацией гена GM2A. Характеризуется нормальной активностью β-гексозаминидаз А и Б и обусловлено недостаточностью активатора (белкового кофактора), необходимого для реализации ферментной активности в отношении субстрата. Заболевание клинически проявляется прогрессирующим разрушением нервных клеток головного и спинного мозга.

Пигментный ретинит (RP) — наследственное, дегенеративное заболевание глаз, которое вызывает сильное ухудшение зрения и часто слепоту. Прогрессирование RP не является последовательным. Некоторые люди могут иметь симптомы с детства, другие могут заметить симптомы позднее. В общем, чем позже начало, тем быстрее ухудшение зрения. Те, кто не имеют RP имеют 90-градусное периферийное зрение, в то время как люди, имеющие RP имеют периферийное зрение менее 90 градусов.
Друзы оптического диска (ODD) или друзы глазного нерва (ONHD) —глобулы из мукопротеинов и мукополисахаридов, которые постепенно кальцифицируют диск зрительного нерва. Они могут стать остатками транспортной системы аксонов вырождающихся ганглиозных клеток сетчатки. ODD также приписывались такие проблемы как врождённые поднятые или аномальные диски, псевдопапиллоэдема, псевдоневрит, утопление друз диска, и гиалиновые тельца в оптическом диске. Они могут быть связаны с потерей зрения различной степени и иногда приводить к слепоте, или, чаще, иметь бессимптомное течение.
Болезнь Штаргардта или жёлтопятнистая абиотрофия сетчатки , является унаследованной формой ювенильной макулярной дегенерации, которая вызывает прогрессирующее ухудшение зрения, как правило, до точки официальной слепоты. Прогрессирование обычно начинается в возрасте от шести до двенадцати лет, и выравнивается вскоре после быстрого снижения остроты зрения. Несколько генов ассоциированы с этим расстройством. Симптомы обычно развиваются до возраста двадцати лет, и включают волнистое видение, слепые пятна, размытость, нарушение цветового зрения, и трудности с адаптацией при тусклом освещении.

Регенерация сетчатки — восстановление утраченных функций сетчатки у позвоночных.

Дистрофия колбочек — наследственное глазное расстройство, характеризующееся потерей колбочек фоторецепторов, ответственных как за центральное, так и цветовое зрение.
Прогрессирующая атрофия сетчатки (PRA) — группа генетических заболеваний, встречающихся у определенных пород собак и, реже, у кошек. Подобно пигментному ретиниту у людей, она характеризуется двусторонней дегенерацией сетчатки, что приводит к прогрессирующей потере зрения, ведущей к слепоте. Болезни этой группы почти во всех породах наследуются как аутосомно-рецессивный признак, за исключением сибирского хаски и бульмастифа. На данный момент не существует лечения от этой группы заболеваний.
В физиологии зрения адаптация — это способность сетчатки глаза приспосабливаться к различным уровням освещённости. Естественное ночное зрение, или скотопическое зрение, — это способность видеть в условиях низкой освещённости. У людей палочки отвечают исключительно за ночное зрение, поскольку колбочки способны функционировать только при более высоких уровнях освещённости. Качество ночного видения ниже, чем дневного, поскольку оно имеет ограниченное разрешение и невозможно различать цвета; видны только оттенки серого. Для того, чтобы перейти от дневного к ночному видению, людям необходимо пройти период темновой адаптации, что может занимать до двух часов. В процессе темновой адаптации каждый глаз приспосабливается от высокого к низкому уровню люминесценции, во много раз повышая чувствительность. Этот период адаптации различается у палочек и колбочек и является результатом регенерации фотопигментов для повышения чувствительности сетчатки. Световая адаптация, напротив, срабатывает очень быстро, за секунды.