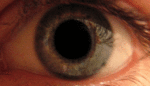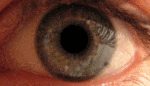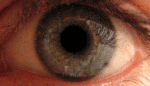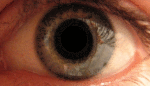
Альбинизм — наследственное заболевание, полное или почти полное отсутствие пигмента меланина или хлорофилла. Проявляется отсутствием нормальной вида окраски кожи, волос, шерсти, радужной и пигментной оболочек глаз, зелёных частей растений.

Сетча́тка — внутренняя оболочка глаза, являющаяся периферическим отделом зрительного анализатора; содержит фоторецепторные клетки, обеспечивающие восприятие и преобразование электромагнитного излучения видимой части спектра в нервные импульсы, а также обеспечивает их первичную обработку.

Макулодистрофи́я — общее название для группы заболеваний, при которых поражается сетчатка глаза и нарушается центральное зрение. В основе макулодистрофии лежит патология сосудов и ишемия центральной зоны сетчатки, которая отвечает за центральное зрение. Возрастная макулодистрофия — одна из самых частых причин слепоты у людей старше 55 лет.

Боле́знь Тея — Са́кса (GM2 ганглиозидоз, ранняя детская амавротическая идиотия) — редкое наследственное заболевание с аутосомно-рецессивным типом наследования, поражающее центральную нервную систему (спинной и головной мозг, а также менингеальные оболочки). Относится к группе лизосомных болезней накопления. Названо в честь британского офтальмолога Уоррена Тея (англ. Warren Tay, 1843—1928) и американского невролога Бернарда Сакса (англ. Bernard Sachs, 1858—1944), которые впервые описали это заболевание независимо друг от друга в 1881 и 1887 годах соответственно.
Лютеин — пигмент, относящийся к ксантофиллам — группе кислородсодержащих каротиноидов. Ксантофиллы — главная составная часть жёлтых пигментов в листьях, цветках, плодах и почках высших растений, а также во многих водорослях и микроорганизмах. Этим термином в 1837 году шведский химик Берцелиус обозначил жёлтый пигмент, выделенный из опавших осенью жёлтых листьев. Позже под ксантофиллами стали понимать только гидроксилированные каротиноиды. Термин «лютеин» используется с XX века. В животном мире ксантофиллы, в том числе лютеин, встречаются реже.
Ранибизумаб — лекарственный препарат, содержащий фрагменты (Fab) моноклональных гуманизированных антител к эндотелиальному фактору роста А (VEGF-A), предназначенный для лечения возрастной макулодистрофии (ВМД) и пролиферативной диабетической ретинопатии (ПДР), разработанный компанией Genentech. В США он распространяется компанией Genentech, в других странах — компанией Novartis под наименованием Луцентис. В настоящее время применение ранибизумаба разрешено более чем в 85 странах для лечения влажной формы ВМД и диабетической ретинопатии.

Макулярный отёк (эдема) происходит, когда жидкость и белковые отложения накапливаются на макуле или под макулой глаза и заставляет её утолщаться и набухать(отёк). Набухание может привести к искажению центрального поля зрения человека, так как пятно располагается рядом с центром сетчатки в задней части глазного яблока. Эта область содержит набор плотно упакованных колбочек, которые обеспечивают резкое, четкое центральное поле зрение, чтобы дать возможность человеку увидеть детали, форму, и цвет объекта, который находится непосредственно в направлении взгляда.
Боле́знь Бильшо́вского — Янско́го — поздняя инфантильная (детская) форма восковидного липофусциноза нейронов, которая развивается на фоне дефицита лизосомного фермента трипептидил-пептидазы-1. Относится к группе лизосомных болезней накопления.
Ретиношизис — глазное заболевание, характеризующееся аномальным расщеплением нейросенсорных слоев сетчатки, как правило, в наружном плексиформном слое. Наиболее распространенные формы являются бессимптомными, некоторые редкие формы приводят к потере зрения в соответствующем поле.
Кристаллическая дистрофия Bietti (BCD), также называемая кристаллическая корнеоретинальная дистрофия Bietti , является редким аутосомно — рецессивным заболеванием глаз и названа в честь доктора G.B. Bietti.

Пигментный ретинит (RP) — наследственное, дегенеративное заболевание глаз, которое вызывает сильное ухудшение зрения и часто слепоту. Прогрессирование RP не является последовательным. Некоторые люди могут иметь симптомы с детства, другие могут заметить симптомы позднее. В общем, чем позже начало, тем быстрее ухудшение зрения. Те, кто не имеют RP имеют 90-градусное периферийное зрение, в то время как люди, имеющие RP имеют периферийное зрение менее 90 градусов.

Периферин 2 представляет собой белок, который у человека кодируется геном PRPH2. Периферин 2 находится в клетках палочек и колбочек сетчатки глаза. Дефекты этого белка являются причиной одной из форм пигментного ретинита, неизлечимой слепоты.
Доминантная атрофия зрительного нерва , или доминантная атрофия зрительного нерва типа Кьер — аутосомное наследственное заболевание, которое влияет на зрительный нерв, что приводит к снижению остроты зрения и слепоте, начиная с детства. Это обусловлено митохондриальной дисфункцией, опосредующей смерть волокон зрительного нерва. Доминантная атрофия зрительного нерва была впервые описана клинически Баттеном в 1896 году и названа оптической нейропатией Кьера в 1959 году в честь датского офтальмолога Poul Kjer, который исследовал 19 семей с болезнью. Хотя домининантная атрофия зрительного нерва является наиболее распространённой аутосомной наследственной оптической нейропатией и не связана с глаукомой, последняя часто ошибочно диагностируется вместо Кьер.
Болезнь Огучи , которая также называется врожденной ночной слепотой , является аутосомно-рецессивной формой врождённой ночной слепоты, связанной с обесцвечиванием глазного дна и аномально медленной адаптацией к темноте.

Бета-маннозидоз — редкое аутосомно-рецессивное наследственное заболевание из группы лизосомных болезней накопления, связанное с нарушением метаболизма олигосахаридов в результате снижения активности фермента лизосом бета-маннозидазы.
Болезнь Штаргардта или жёлтопятнистая абиотрофия сетчатки , является унаследованной формой ювенильной макулярной дегенерации, которая вызывает прогрессирующее ухудшение зрения, как правило, до точки официальной слепоты. Прогрессирование обычно начинается в возрасте от шести до двенадцати лет, и выравнивается вскоре после быстрого снижения остроты зрения. Несколько генов ассоциированы с этим расстройством. Симптомы обычно развиваются до возраста двадцати лет, и включают волнистое видение, слепые пятна, размытость, нарушение цветового зрения, и трудности с адаптацией при тусклом освещении.

Член 4 подсемейства A (АВС1) ATP-связывающей кассеты , также известный как ABCA4 или ABCR — белок, который в организме человека кодируется геном ABCA4.

Дистрофия колбочек — наследственное глазное расстройство, характеризующееся потерей колбочек фоторецепторов, ответственных как за центральное, так и цветовое зрение.

Болезнь Шарко — Мари — Тута (ШМТ), или наследственная моторно-сенсорная нейропатия (НМСН) — наследственная периферическая нейропатия с хроническим прогрессирующим течением. При этой болезни больные страдают от слабости и атрофии мышц дистальных отделов конечностей, деформации стоп и кистей, у них наблюдается снижение сухожильных рефлексов, изменение походки, потеря чувствительности в конечностях. В основе клинических проявлений болезни лежит поражение двигательных и чувствительных периферических нервных волокон. Болезнь Шарко — Мари — Тута диагностируется с приблизительной частотой 1 на 2500 человек. Первое проявление болезни чаще всего происходит в подростковом возрасте или в раннем взрослом возрасте. Тяжесть симптомов сильно различается даже среди членов одной семьи с этим заболеванием. Болезнь Шарко — Мари — Тута нередко ведёт к ограничению трудоспособности и к инвалидизации, при этом большинство пациентов имеют нормальную продолжительность жизни. Болезнь Шарко — Мари — Тута является генетически крайне неоднородным заболеванием, симптомы этой болезни могут быть вызваны мутациями в более чем двух десятках генов, хотя большая часть заболеваний вызвана мутациями в генах PMP22, MPZ, GJB1 и MFN2. Наследование болезни чаще всего аутосомно-доминантное, однако может быть аутосомно-рецессивным и Х-сцепленным.
Прогрессирующая атрофия сетчатки (PRA) — группа генетических заболеваний, встречающихся у определенных пород собак и, реже, у кошек. Подобно пигментному ретиниту у людей, она характеризуется двусторонней дегенерацией сетчатки, что приводит к прогрессирующей потере зрения, ведущей к слепоте. Болезни этой группы почти во всех породах наследуются как аутосомно-рецессивный признак, за исключением сибирского хаски и бульмастифа. На данный момент не существует лечения от этой группы заболеваний.