Витили́го ; арх. песь — нарушение пигментации, выражающееся в исчезновении пигмента меланина на отдельных участках кожи. Предрасположенность к витилиго может наследоваться. Природа заболевания до конца не изучена.
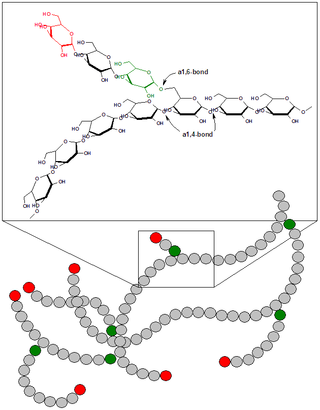
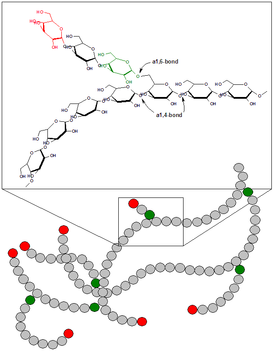
Болезнь Андерсена — гликогеноз, семейный цирроз печени, вызванный дефектом фермента амило-(1,4-1,6)-трансглюкозилазы.

Болезнь Мак-Ардля — гликогеноз, связанный с дефектом мышечной фосфорилазы.
Боле́знь Герса — гепатофосфорилазная недостаточность, гликогеноз, вызванный недостаточностью фосфорилазы печени.
Ме́тгемоглобинеми́я — состояние, характеризующееся появлением в крови повышенного количества метгемоглобина.
Гликоге́н — гексоза, гомополисахарид состава (C6H10O5)n, образованный остатками глюкозы, соединёнными связями α-1→4 (в местах разветвления — α-1→6). В клетках животных служит основным запасным углеводом и основной формой хранения глюкозы. Откладывается в виде гранул в цитоплазме в клетках многих типов (главным образом в клетках печени и мышц).

Ге́рти Тере́за Ко́ри, урождённая Ра́дниц — американский биохимик. Член Национальной академии наук США (1948).

Ту́булопати́и — большая группа заболеваний, протекающих с нарушением канальцевого транспорта органических веществ и электролитов.

Мукополисахаридо́зы сокращённо МПС, или англ. MPS — группа метаболических заболеваний соединительной ткани, связанных с нарушением обмена кислых гликозаминогликанов, связанных недостаточностью лизосомных ферментов обмена гликозаминогликанов. Заболевания вызваны наследственными аномалиями обмена, проявляются в виде лизосомной болезни накопления: различных дефектов костной, хрящевой, соединительной тканей.
Концентрический склероз — хроническое быстро прогрессирующее демиелинизирующее заболевание, развивается преимущественно у относительно молодых пациентов. Ряд авторов рассматривают его, также как и болезнь Девика, не как отдельную нозологическую единицу, а как один из вариантов рассеянного склероза, хотя в МКБ-10 они кодируются в самостоятельных подрубриках.
Паренхимато́зные дистрофи́и — нарушения метаболизма в паренхиме органов.
Углеводный обмен, или метаболизм углеводов в организмах животных и человека. Метаболизм углеводов в организме человека состоит из следующих процессов:
- Расщепление в пищеварительном тракте поступающих с пищей поли- и дисахаридов до моносахаридов, дальнейшее всасывание моносахаридов из кишечника в кровь.
- Синтез и распад гликогена в тканях, прежде всего в печени.
- Гликолиз — распад глюкозы. Первоначально под этим термином обозначали только анаэробное брожение, которое завершается образованием молочной кислоты (лактата) или этанола и углекислого газа. В настоящее время понятие «гликолиз» используется более широко для описания распада глюкозы, проходящего через образование глюкозо-6-фосфата, фруктозо-1,6-дифосфата и пирувата как в отсутствие, так и в присутствии кислорода. В последнем случае употребляется термин «аэробный гликолиз», в отличие от «анаэробного гликолиза», завершающегося образованием молочной кислоты или лактата.
- Анаэробный путь прямого окисления глюкозы или, как его называют, пентозофосфатный путь.
- Взаимопревращение гексоз.
- Анаэробный метаболизм пирувата. Этот процесс выходит за рамки углеводного обмена, однако может рассматриваться как завершающая его стадия: окисление продукта гликолиза — пирувата.
- Глюконеогенез — образование углеводов из неуглеводных продуктов.

Лизосо́мные боле́зни накопле́ния — общее название группы весьма редких наследственных заболеваний, вызванных нарушением функции внутриклеточных органелл лизосом. Эти одномембранные органоиды являются частью эндомембранной системы клетки и специализируются на внутриклеточном расщеплении веществ: гликогена, гликозаминогликанов, гликопротеинов и других. Лизосомные болезни накопления вызываются генетически обусловленным дефицитом ферментов лизосом, что приводит к накоплению макромолекул, являющихся субстратом этих ферментов, в различных органах и тканях организма.

Боле́знь По́мпе — редкое, наследственное заболевание с аутосомно-рецессивным механизмом, наследования,, связанное с повреждением мышечных, и нервных клеток по всему организму. Клиническая картина данной патологии, обусловлена накоплением гликогена, в лизосомах, вызванным недостаточностью лизосомного фермента — кислой α-1,4-глюкозидазы. Различают быстро прогрессирующую (классическую) и медленно прогрессирующую формы болезни Помпе. Несмотря на широкий спектр клинических проявлений, гликогеноза II типа, в основе всех форм болезни лежит, дефицит одного фермента, кодируемого геном GAA.
Недоста́точность ки́слой фосфата́зы — редкое наследственное заболевание из группы лизосомных болезней накопления. Клиническая симптоматика дефицита кислой фосфатазы развивается в раннем возрасте. Характеризуется гепатомегалией и задержкой психического развития.

Бета-маннозидоз — редкое аутосомно-рецессивное наследственное заболевание из группы лизосомных болезней накопления, связанное с нарушением метаболизма олигосахаридов в результате снижения активности фермента лизосом бета-маннозидазы.

Фукозидо́з — редкое наследственное заболевание из группы лизосомных болезней накопления, связанное с недостаточностью гидролаз, расщепляющих полисахаридные связи. В результате внутри клеток накапливаются два класса макромолекул: гликолипиды и гликопротеины. Клиническая картина связана с мутацией, приводящей к повреждению или дефициту фермента лизосом альфа-L-фукозидазы и характеризуется разнообразными соматическими проявлениями и нарушением функции нервной системы.
Анри́-Жери́ Э́рс — бельгийский физиолог и биохимик, профессор Лувенского католического университета.
Альфа-маннозидо́з — редкое аутосомно-рецессивное наследственное заболевание из группы лизосомных болезней накопления, связанное с нарушением метаболизма олигосахаридов в результате снижения активности фермента лизосом альфа-маннозидазы. Также заболевание наносит ущерб в животноводстве.
Болезнь Хага — гликогеноз 9 типа. Данное заболевание относится к группе гликогеновых болезней. Причиной является мутация гена PHKA2, отвечающего за синтез α2 -субъединицы фермента киназы фосфорилазы. Этот ген располагается на коротком плече 10-й хромосомы в области Хр22.13. Данное заболевание относится к печеночной форме гликогенозов, то есть происходит нарушение использования гликогена, который необходим для поддержания нормального уровня глюкозы в крови. Болезнь описана врачом Г. Хагом.