Инфракрасная спектроскопия

Инфракра́сная спектроскопи́я (колебательная спектроскопия, средняя инфракрасная спектроскопия, ИК-спектроскопия, ИКС) — раздел спектроскопии, изучающий взаимодействие инфракрасного излучения с веществами.
При пропускании инфракрасного излучения через вещество происходит возбуждение колебательных движений молекул или их отдельных фрагментов. При этом наблюдается ослабление интенсивности излучения, прошедшего через образец. Однако поглощение происходит не во всём спектре падающего излучения, а лишь при тех длинах волн, энергия которых соответствует энергиям возбуждения колебаний в изучаемых молекулах. Следовательно, длины волн (или частоты), при которых наблюдается максимальное поглощение ИК-излучения, могут свидетельствовать о наличии в молекулах образца тех или иных функциональных групп и других фрагментов, что широко используется в различных областях химии для установления структуры соединений.
Экспериментальным результатом в ИК-спектроскопии является инфракрасный спектр — функция интенсивности пропущенного инфракрасного излучения от его частоты. Обычно инфракрасный спектр содержит ряд полос поглощения, по положению и относительной интенсивности которых делается вывод о строении изучаемого образца. Такой подход стал возможен благодаря большому количеству накопленной экспериментальной информации: существуют специальные таблицы, связывающие частоты поглощения с наличием в образце определённых молекулярных фрагментов. Созданы также базы ИК-спектров некоторых классов соединений, которые позволяют автоматически сравнивать спектр неизвестного анализируемого вещества с уже известными и таким образом идентифицировать это вещество.
Инфракрасная спектроскопия является ценным аналитическим методом и служит для исследования строения органических молекул, неорганических и координационных, а также высокомолекулярных соединений. Основным прибором, используемым для подобных анализов, является инфракрасный спектрометр (дисперсионный или с преобразованием Фурье).
Анализ сложных образцов стал возможен благодаря разработке новых техник инфракрасной спектроскопии: ИК-спектроскопии отражения, ИК-спектроскопии испускания и ИК-микроскопии. Кроме того инфракрасная спектроскопия была объединена с другими аналитическими методами: газовой хроматографией и термогравиметрией.
История метода

Инфракрасное излучение было открыто в 1800 году астрономом Уильямом Гершелем. Используя призму, он наблюдал повышение температуры в области, находящейся за красной границей спектра видимого излучения. В 1882—1900 годах Уильям Эбни и Эдвард Фестинг[англ.] записали инфракрасные спектры 52 соединений и сопоставили наблюдаемые полосы поглощения с функциональными группами, присутствующими в этих молекулах. Существенный вклад в метод сделал американский физик Уильям Кобленц, который с 1903 года, пользуясь призмой из хлорида натрия, получил весьма точные и полные ИК-спектры для сотен органических и неорганических веществ[1][2].
Первые эксперименты по регистрации инфракрасных спектров были крайне трудоёмкими, поскольку исследователи были вынуждены собирать собственные приборы, шлифовать и полировать призмы, серебрить зеркала, градуировать приборы по показателям преломления каменной соли. При этом спектрометры были чувствительны к вибрациям, поэтому их располагали на фундаменте, а исследования проводили по ночам. Время регистрации одного спектра составляло от 3 до 4 часов. Уже в ранних работах было показано, что ИК-спектры соединений имеют индивидуальный вид[1].
В то время природа поглощения инфракрасного излучения не была до конца ясна, однако к 1930-м годам была создана теория, в которой полагалось, что это поглощение наблюдается вследствие колебаний молекул и что характер этого поглощения так или иначе связан с изменением дипольного момента, правилами отбора, симметрией молекул и степенью ангармонизма колебаний[2].
В 1940 году фирмы Dow Chemical и American Cyanamid[англ.] создали собственные однолучевые приборы для изучения углеводородов. Коммерческие спектрометры стали выпускаться в 1946 году при сотрудничестве American Cyanamid с Perkin-Elmer[англ.]. Доступность приборов привела к созданию обширных таблиц корреляции наблюдаемых полос поглощения со структурой поглощающих функциональных групп[3].
После Второй мировой войны появилась возможность усиливать слабый сигнал ИК-спектрометров, что сократило время эксперимента до 1—2 часов. Затем была усовершенствована техника изготовления термоэлектрических приёмников с малым временем отклика. Эти улучшенные детекторы позволяли избежать дрейфа показаний во времени и привели к созданию двухлучевых приборов, где шкала калибровалась в процентах пропускания против шкалы длин волн или волновых чисел[1].
Стало возможным промышленное получение больших и качественных кристаллов галогенидов щелочных металлов, необходимых для создания оптических элементов приборов, что позволило преодолеть многие трудности. Например, синтетический бромид калия, в отличие от использовавшейся ранее каменной соли, позволил записывать ИК-спектры вплоть до 400 см−1, тогда как прежний предел составлял 650 см−1[4].
Расцветом ИК-спектроскопии стало появление ИК-интерферометров, которые первоначально использовались для детектирования очень слабого инфракрасного излучения астрономических объектов. После разработки быстрых методов конвертирования интерферограмм в спектры (преобразование Фурье) и уменьшения времени сканирования подобные приборы стали выпускаться серийно, что в 1970-е годы позволило выйти на рынок ИК-спектрометров компаниям, которые производили компьютеры, но не имели опыта работы в области спектроскопии (Nicolet, Bruker). Преимущество ИК-интерферометров заключалось в их мультиплексности (преимущество Фелгетта), то есть одновременном сборе информации о поглощении всех длин волн, за счёт чего достигалось более высокое отношение сигнала к шуму для фиксированного времени сканирования спектра. Второе преимущество состояло в производительности приборов нового типа: в то время как дисперсионные приборы имели вход и выход, которые ограничивали количество проходящего через них света, производительность интерферометра определялась толщиной пучка света от источника. Вероятно, мода также сыграла значительную роль в распространении ИК-спектрометров с преобразованием Фурье, поскольку в то время не существовало большой необходимости в высоком соотношении сигнал/шум: образцы обычно готовились гораздо дольше, чем проводилось измерение, и масса образцов была достаточной для записи качественных спектров[5].
ИК-интерферометры позволили получать спектры в дальней ИК-области, наблюдать решёточные колебания кристаллов, а также, благодаря высокому отношению сигнал/шум, преодолевать сложности при интерпретации спектров органических соединений. Одним из популярных занятий в то время была цифровая обработка спектров, а именно удаление полос поглощения растворителей, определение степени чистоты и характера примесей. Интерферометры нашли широкое применение в исследовании водных растворов биологических молекул[6].
В 1980-е годы появились комбинированные методы, объединившие газовую хроматографию и ИК-спектроскопию. Напольные приборы большого размера сменились более компактными настольными моделями. Появилась возможность ступенчатого сканирования во времени, что позволило изучать динамические процессы со сбором данных в одной точке[6].
Принцип метода
Основные характеристики ИК-излучения

ИК-спектроскопия основана на явлении поглощения химическими веществами инфракрасного излучения с одновременным возбуждением колебаний молекул. Инфракрасное излучение представляет собой электромагнитную волну и характеризуется длиной волны λ, частотой и волновым числом , которые связаны следующей зависимостью:



где с — скорость света, а n — показатель преломления среды[7].
В спектроскопии поглощения, частным случаем которой является ИК-спектроскопия, происходит поглощение молекулами фотонов определённой энергии, которая связана с частотой электромагнитной волны через постоянную Планка:

При поглощении фотона происходит возбуждение — увеличение энергии молекулы: она переходит из основного колебательного состояния E1 в некоторое возбуждённое колебательное состояние E2 так, что энергетическая разница между этими уровнями равна энергии фотона[7].

Энергия поглощённого инфракрасного излучения расходуется на возбуждение колебательных переходов для веществ в конденсированном состоянии. Для газов поглощение кванта ИК-излучения приводит к колебательным и вращательным переходам[7].
Виды и энергия колебаний молекул
Колебательные движения молекул определяются их внутренними, или колебательными, степенями свободы. Число колебательных степеней свободы и соответствующих им нормальных[K 1] колебаний равно (3n-5) для линейных молекул и (3n-6) для нелинейных молекул, где n — число атомов в молекуле[K 2]. Например, молекула воды H2O нелинейна и имеет 3 колебательные степени свободы, а линейная молекула водорода H2 — лишь одну[8][9].
Колебания молекул могут заключаться в изменении длин связей (валентные колебания, v) либо углов между связями (деформационные колебания, δ). Валентные колебания могут быть симметричными и антисимметричными, а деформационные колебания подразделяются на ножничные, маятниковые, веерные и крутильные. Для более сложных молекул, в которых одна из деформационно колеблющихся частей гораздо массивнее другой, деформационные колебания чаще описывают как плоскостные и внеплоскостные. Колебания, которые заключаются в одновременном изменении нескольких длин связей или валентных углов, называются скелетными[10].
| Валентные колебания (Stretching) | Деформационные колебания | ||||
|---|---|---|---|---|---|
| симметричное | антисимметричное | плоскостные | внеплоскостные | ||
| ножничное (scissoring) | маятниковое (rocking) | веерное (wagging) | крутильное (twisting) | ||
 |  |  |  |  |  |

Колебания молекул могут быть описаны при помощи моделей гармонического и ангармонического осциллятора. С точки зрения модели гармонического осциллятора, двухатомная молекула представляет собой две массы m1 и m2, соединённые упругой пружиной, не имеющей массы, с силовой постоянной K. В таком случае частота колебания атомов такой молекулы вдоль линии, проходящей через центры их масс, равна[11]:


Из данных выражений следует, что наблюдаемая частота колебаний двухатомного осциллятора зависит от силовой постоянной K, которая, в свою очередь, связана с энергией связи между двумя атомами, а также от массы атомов, участвующих в колебании. Для многоатомных молекул колебания носят более сложный характер и приближение гармонического осциллятора неприменимо[11].
Потенциальная энергия гармонического осциллятора связана с отклонением расстояния между атомами X следующим образом[11]:

График потенциальной энергии представляет собой параболу, симметричную относительно начального положения атомов в состоянии покоя (re). Согласно квантовой механике, энергетические состояния молекулы квантованы, то есть являются дискретными. Подобные квантованные состояния называют колебательными уровнями. Колебательные уровни отстоят друг от друга на одинаковом расстоянии, и их энергия может быть вычислена по уравнению[11]

При vi = 0 молекула находится на самом низком колебательном уровне, и колебательная энергия в таком состоянии равна E = ½ hν. Данная энергия всегда присуща молекуле и не может быть отобрана. В приближении гармонического осциллятора разрешены лишь переходы с Δv = ±1, то есть лишь на соседние уровни (правило отбора)[11].
Более точной является модель ангармонического осциллятора. Ангармоничность проявляется, если величина дипольного момента изменяется не пропорционально смещению атомов. Отличие данной модели состоит в том, что расстояние между колебательными уровнями уменьшается с увеличением номера уровня. Отклонение от гармоничности также увеличивается снизу вверх. Энергия уровня в случае ангармонического осциллятора выражается следующим образом[11]:

Ангармоничность колебаний приводит к уменьшению строгости правила отбора, вследствие чего в спектрах могут наблюдаться переходы с Δv = ±2 — обертоны. Как правило, частота обертона попадает в область 2×ν1-b, где b = 2—10 см−1. Возможно также возникновение комбинационных, или составных, полос, имеющих частоту ν1 + ν2, где ν1 и ν2 — частоты каких-либо фундаментальных колебаний молекулы. Комбинационная полоса появляется при колебательных переходах из возбуждённых состояний. Обычно для конденсированного состояния интенсивность обертонов и комбинационных полос в 10—100 раз ниже, чем основных, хотя могут встречаться исключения[12].
Если обертон или комбинационная полоса совпадают по частоте с каким-либо фундаментальным колебанием, проявляется резонанс Ферми, который приводит к появлению двух полос поглощения примерно одинаковой интенсивности, в то время как ожидается наличие лишь одной фундаментальной полосы. Иногда также происходит смешивание колебаний с примерно одинаковой частотой: при этом число колебаний остаётся таким же, но они проявляются при других частотах и уже не могут быть отнесены только к одной связи. Осложняющим фактором также является появление в спектрах тонкой структуры, соответствующей вращательным переходам (такое явление наблюдается лишь для веществ в газообразном состоянии)[10].
Характеристические колебания
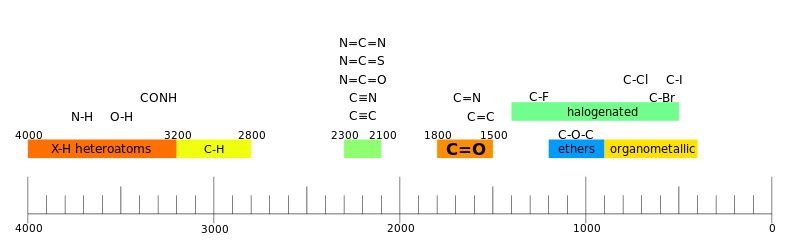
Многоатомные молекулы имеют 3n-6(5) нормальных колебаний, и в каждом таком колебании участвуют не пары атомов при одной связи, а в той или иной степени все n атомов молекулы. Однако было экспериментально установлено, что для колебаний некоторых функциональных групп вклад «посторонних» атомов и связей достаточно мал, поэтому вне зависимости от окружения эти функциональные группы поглощают в ограниченном интервале частот. Этот факт позволил путём сравнения многочисленных спектров соотнести наличие в молекуле характерных фрагментов с наблюдаемыми полосами поглощения. Такие полосы получили название групповых, или характеристических. По ним можно быстро и однозначно подтвердить присутствие или отсутствие в молекуле соответствующих фрагментов[13].
Возникновение характеристических колебаний может происходить по двум причинам[14]:
- Если характеристическое колебание относится к лёгкому атому, связанному с тяжёлым, то практически всё движение сосредоточено именно на нём, и влияние остальной части молекулы на него весьма слабое.
- Колебания, относящиеся к атомам очень близкой массы (например, C=O, C≡N), слабо взаимодействуют с колебаниями остальных частей молекулы.
Существуют также менее определённые характеристические колебания, которые наблюдаются в сравнительно более широком интервале частот. Однако их положение в спектре можно объяснить массой атомов, резонансом или электронными эффектами в молекуле[14].
Поглощение излучения
Обычно в эксперименте прибор испускает одновременно все длины волн инфракрасного излучения, включая ближнюю ИК-область (14 000 — 4000 см−1), среднюю ИК-область (4000 — 400 см−1) и дальнюю ИК-область (400 — 10 см−1). Поглощение излучения веществом количественно описывается законом Бугера — Ламберта — Бера, а спектр получается при построении зависимости пропускания (T, англ. transmittance, %) или оптической плотности (D, англ. optical density) от длины волны (частоты, волнового числа)[15].
Для того, чтобы поглощение излучения произошло, необходимо выполнение двух условий. Во-первых, поглощаются лишь волны такой частоты, которая совпадает с частотой того или иного колебания молекулы. Во-вторых, колебание должно вызывать изменение дипольного момента молекулы. По этой причине молекулы, не имеющие дипольного момента (например, H2, N2, O2, а также соли без ковалентных связей и металлы), не поглощают инфракрасное излучение. Интенсивность полос в ИК-спектре пропорциональна квадрату изменения дипольного момента[15][16].
ИК-спектрометры
Дисперсионные ИК-спектрометры
В дисперсионных ИК-спектрометрах роль монохроматора может выполнять призма либо — в более новых моделях приборов — дифракционная решётка. Обычно в оптической схеме монохроматор располагается после кюветы с анализируемым веществом, то есть в спектр разлагается излучение, взаимодействовавшее с образцом. При этом последовательно для каждой длины волны излучения регистрируется интенсивность излучения, что и даёт спектр поглощения. На пути излучения установлена щель регулируемой ширины, позволяющая выделить для работы определённый спектральный интервал (обычно от 20 до 0,5 см−1)[17].
Наиболее часто используются двухлучевые дисперсионные ИК-спектрометры. В этом случае излучение источника делится на две части, одна из которых пропускается через анализируемый образец, а вторая — через образец сравнения (чистый растворитель, или таблетка бромида калия без пробы). Эти два пучка попеременно попадают на детектор, где создают сигналы разной интенсивности. Их соотношение даёт величину пропускания Т[17].
Спектрометры с преобразованием Фурье
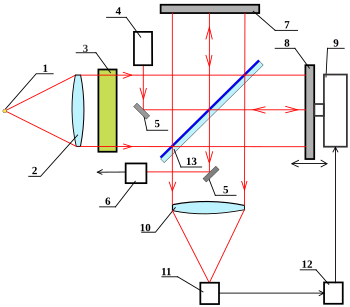
Фурье-спектрометр представляет собой интерферометр Майкельсона, в котором одно из зеркал выполнено подвижным, что позволяет варьировать разницу хода лучей. Смещение зеркала производится механическим приводом, управляемым ЭВМ.
1 — Источник белого света или исследуемый источник;
2 — Линза коллиматора;
3 — Кювета с исследуемым веществом;
4 — Опорный (эталонный) лазер;
5 — Вспомогательные зеркала опорного пучка от лазера;
6 — Фотоприёмник опорного пучка;
7 — Неподвижное зеркало;
8 — Подвижное зеркало;
9 — Механический привод подвижного зеркала;
10 — Объектив фотоприёмника;
11 — Фотоприёмник;
12 — Управляющий и обрабатывающий интерферограмму компьютер;
13 — Светоделительная пластина.

Главным компонентом Фурье-ИК-спектрометров является интерферометр Майкельсона, известный с конца 19-го века. Его ключевыми элементами являются три зеркала. Светоделительное зеркало (пластина) делит пучок излучения на две части, одна из которых отражается от неподвижного зеркала, а вторая — от подвижного (сканера). Оба отражённых пучка затем снова попадают на светоделительное зеркало, где объединяются и направляются на детектор (фотоприёмник). Подвижное зеркало призвано создавать разницу оптического пути (разность хода) для двух пучков света. При разности хода в проходящие пучки взаимно уничтожаются, а отражённые, напротив, усиливаются. В результате получается интерферограмма — график зависимости интенсивности зарегистрированного излучения от разности хода пучков. Для монохроматического света она имеет форму косинусоиды. Для используемого в ИК-спектроскопии полихроматического света она приобретает более сложную форму и содержит всю спектральную информацию о падающем на детектор пучке. Далее интерферограмма пересчитывается в инфракрасный спектр путём преобразования Фурье[18][19].

Преимущество таких приборов заключается в следующем:[20]
- одновременно регистрируются все длины волн;
- на детектор попадает более интенсивный поток света за счёт отсутствия щелей;
- в качестве внутреннего эталона длины волны используется гелий-неоновый лазер;
- возможна запись спектров в режиме накопления.
Как следствие, значительно сокращается время записи спектра: спектрометры с преобразованием Фурье дают возможность записать до 50 спектров за секунду, в то время как дисперсионный прибор требует около 20 минут для записи одного спектра. Также улучшается качество спектров и чувствительность анализа (на 2-3 порядка) за счёт использования режима накопления[K 3]. Фурье-ИК-спектрометры обычно однолучевые, что делает невозможным запись спектра с образцом сравнения. По этой причине также не удаётся компенсировать «атмосферные» помехи (наличие углекислого газа и воды). Обычно этот недостаток устраняется путём записи двух последовательных спектров с вычитанием спектра образца сравнения из спектра анализируемого образца, однако в последнее время также приобретают популярность двухлучевые приборы[18].
ИК-спектроскопия пропускания
Органические соединения

Колебательные спектры органических соединений обычно имеют сложную структуру и содержат большое число полос разной формы и интенсивности. Экспериментально установлено, что наличие тех или иных полос в определённой области спектра свидетельствует о наличии в молекуле соответствующих им функциональных групп. Однако ни одна группа не является в полной мере изолированной от колебаний остальной части молекулы. Это приводит к некоторым изменениям частоты и интенсивности полос, зависящим от химического окружения функциональной группы[21].
Анализ ИК-спектров многих тысяч органических соединений позволил составить корреляционные таблицы, которые связывают функциональные группы с частотой и интенсивностью колебаний. Однако обычно в спектрах органических соединений присутствуют также полосы поглощения, которые нельзя соотнести с конкретными колебаниями[21].
Колебания связей X-H, где X: C, O или N, можно приближённо описать, как колебания двухатомной молекулы. В этом случае приведённая масса μ всегда близка к 1, а значение силовой постоянной K примерно одинаково для всех подобных связей, поэтому колебания X-H проявляются приблизительно в одной области частот. Например, для связи C-H силовая постоянная равна около 490 Н/м, что даёт частоту в 3000 см−1. Для связей O-H и N-H значение частоты обычно немного выше из-за более высоких значений K[21].
Для связей X—X' приведённая масса гораздо выше, например, для связи C-O она составляет 6,86. Поскольку силовая постоянная примерно такая же, как для связи C-H (обе связи одинарные), то частота колебаний C-O должна быть в √6,86 раз ниже, чем 3000 см−1, то есть 1150 см−1. Аналогичные соображения применимы для кратных связей. Например, силовая постоянная связи C=O примерно в два раза выше, чем для связи C-O, соответственно, оценочная частота её колебания составляет 1600 см−1 (реальное усреднённое значение равно 1700 см−1). Тройные связи прочнее двойных, и их колебания наблюдаются в диапазоне 2300—2100 см−1[21].
Силовые постоянные для деформационных колебаний ниже, чем для соответствующих валентных колебаний, поэтому они проявляются при более низких частотах. Например, колебания групп XH2 попадают в область 1500 см−1, групп XYH — в область от 1500 до 1000 см−1, а групп XYZ (все атомы относительно тяжёлые) — в область ниже 1000 см−1[21].
Таким образом, спектр можно разделить на четыре области:
- 3600—2800 см−1 — область валентных колебаний X-H;
- 2800—1800 см−1 — область колебаний тройных связей либо других относительно редких групп;
- 1800—1500 см−1 — область колебаний двойных связей;
- ниже 1500 см−1 — область отпечатков пальцев[21].
Изотопное замещение приводит к тому, что частота колебания смещается, причём экспериментально наблюдаемый сдвиг обычно хорошо согласуется с расчётным. Теоретически, замена атома водорода на дейтерий должна привести к изменению частоты в √2 = 1,414 раза, но на практике это смещение зависит также от типа колебания, например, для валентного симметричного и маятникового колебаний коэффициент изменения составляет 1,379 раза, для валентного асимметричного — 1,349 раза, для плоскостного деформационного — 1,349 раза, для веерного — 1,323 раза, и для крутильного — 1,414 раза. Также отклонение от расчётных параметров наблюдается из-за небольшого укорочения длины связи при замещении более тяжёлым изотопом, например, для молекул H35Cl и D35Cl равновесные расстояния между центрами атомов равны 1,2837 и 1,2813 Å[K 4][22].
Неорганические, координационные и металлоорганические соединения

Вид инфракрасного спектра неорганического соединения зависит прежде всего от его агрегатного состояния. Для газообразных образцов возможно проявление тонкой вращательной структуры вследствие перехода между вращательными состояниями. При переходе к жидким образцам вращательные переходы исчезают из-за высокой частоты столкновений молекул в растворах. Также в конденсированных веществах возникают новые полосы поглощения ниже при частотах ниже 300 см−1, которые соответствуют решёточным колебаниям[23].
В области ближнего ИК-излучения (12 500—4000 см−1) обычно проявляется много полос, соответствующих обертонам фундаментальных или составных колебаний. Для среднего ИК-излучения выделяют область групповых частот (4000—1300 см−1) и область отпечатков пальцев (1300—650 см−1). В первой области проявляются групповые колебания, приписываемые каким-либо па́рам атомов молекулы: от 4000 до 2500 см−1 — колебания с участием атомов водорода, от 2500 до 2000 см−1 — колебания тройных связей, от 2000 до 1540 см−1 — колебания двойных связей. В области отпечатков пальцев находятся деформационные и скелетные колебания многоатомных систем[23].
Область дальнего ИК-излучения (от 667 до 10 см−1) содержит деформационные колебания углерода, азота, кислорода и фтора, которые связаны с атомами с массой выше 19 а. е. м., а также деформационные колебания циклических и ненасыщенных систем. Колебания в данной области особенно чувствительны к химическому окружению и могут быть использованы для установления строения изомеров. Также данная область полезна при изучении металлоорганических соединений, поскольку частоты колебаний сильно зависят от комплексообразующего атома[23].
Высокомолекулярные соединения

В отличие от низкомолекулярных соединений, полимеры состоят из длинных цепей. Это приводит к тому, что для них можно наблюдать дополнительные полосы поглощения, которые относятся к колебаниям целых цепей[24].
Получение ИК-спектров для высокомолекулярных соединений затруднено тем, что они сильно поглощают ИК-излучение. Для того, чтобы величина оптической плотности попадала в допустимые пределы, согласно закону Бугера — Ламберта — Бера, необходимо, чтобы толщина полимера составляла около 5 мкм. Получить материал такой толщины можно в лаборатории, однако распространённые материалы гораздо толще, поэтому для анализа полимеров используют такие приёмы, как нарушенное полное внутреннее отражение (НПВО) и спектроскопию внешнего отражения. Эффективный оптический путь в этих методах гораздо меньше (1 мкм и меньше)[24].
Обычно структуру полимера устанавливают, применяя совместно методы ИК-спектроскопии и спектроскопии комбинационного рассеяния. Первая даёт информацию о функциональных группах с большим дипольным моментом (C-H, C=O), тогда как вторая чувствительна к поляризуемым группам, содержащимся в макромолекулярных цепях (C-C, C=C). По ИК-спектрам можно определить количество концевых функциональных групп в навеске высокомолекулярного соединения и оценить его молекулярную массу. ИК-спектроскопия также помогает определить составные компоненты в сополимерах, а также низкомолекулярные добавки и примеси. Особенно это важно в промышленности, при идентификации и установлении свойств полимера. При использовании соответствующих баз данных, с которыми машинный алгоритм сравнивает снятый ИК-спектр, эта процедура может выполняться рутинно. По сигналам в ИК-спектре также определяется степень разветвления полимеров и их стереорегулярность, кристалличность либо аморфность полимеров[24].
Подготовка образцов

Регистрация спектров жидких веществ обычно осуществляется из тонких плёнок, расположенных между стёклами из материалов, пропускающих ИК-излучение. Для органических веществ обычно применяется бромид калия. Для водных растворов может применяться селенид цинка, который обладает широким спектральным окном пропускания, однако его высокий коэффициент преломления приводит к появлению полос интерференции, что затрудняет количественное определение. Заменой ему могут служить фторид кальция и фторид бария. Стёкла из галогенидов щелочных металлов быстро мутнеют во влажной атмосфере и легко царапаются, но могут быть легко отполированы заново[25].
Существует три приёма для записи спектров жидкостей: в ячейках с фиксированными или съёмными спейсерами (прокладками между стекол) либо из плёнки непосредственно между стёклами. Для количественных измерений предпочтительно использование фиксированных ячеек, где спейсером служит амальгама, которая обеспечивает постоянный оптический путь. В разборных ячейках эту функцию выполняет прокладка из металлической фольги или тефлона. Стёкла в таких ячейках можно разбирать для очистки или изменения длины оптического пути. Обычно после повторной сборки оптический путь немного изменяется, однако он может быть легко рассчитан вновь по полосам интерференции. Толщина зазора между стёклами пустой ячейки рассчитывается как (2 × расстояние между полосами)−1. Согласно третьему способу, каплю жидкости можно поместить на одно стекло и прижать её другим. При этом оптический путь регулируется прилагаемым усилием. Это удобно для количественных оценок, но неудобно для летучих веществ[25].
Если образец неустойчив или легко испаряются, для записи спектра можно использовать кюветы с водяным охлаждением. Выбор такой кюветы может быть существенным в случае прибора с большой мощностью инфракрасного излучения[26].
Также одним из приёмов для записи спектров жидких веществ является их растворение в подходящем растворителе. Обычно для этой цели не используют воду, поскольку она несовместима со многими оптическими материалами и сильно поглощает в ИК-области. Лучше всего использовать растворители, состоящие из симметричных молекул, так как они дают минимальное число полос в инфракрасном спектре. Особое значение в этом смысле имеют сероуглерод и четырёххлористый углерод[26].
Для получения спектров твёрдых веществ их нужно измельчить в мелкий порошок и диспергировать в матрице. В качестве матрицы обычно используется бромид калия: в количестве 200—300 мг он смешивается с образцом (1-2 мг), растирается (для получения качественных спектров желательно, чтобы размер частиц не превышал длины волны излучения), вакуумируется для удаления воды и прессуется ручным гидравлическим прессом (обычно мощностью 15 или 25 тонн) в небольшую таблетку диаметром 13 мм и толщиной 1 мм. Перед использованием бромид калия можно прогревать до 40 °С для того, чтобы на нём не конденсировалась вода, которая даже в минимальном количестве проявляется в спектре в виде полос при 3450 и 1640 см−1. Преимуществом такого приёма является то, что бромид калия не поглощает в области выше 400 см−1. Также растворимые вещества можно нанести на стекло в виде раствора, после чего удалить растворитель под инфракрасной лампой[25][27].
Альтернативным материалом матрицы служит вазелиновое масло (нуйол[англ.]). Образец в этом случае готовится растиранием в ступке с несколькими каплями такого масла. Получаемая смесь помещается в разборную кювету, после чего записывается спектр. Примесными сигналам от матрицы являются сигналы CH3- и CH2-групп. Наблюдать сигналы в области 3000 см−1 позволяет матрица из фторированных углеводородов (fluorolube)[28].
Если образец представляет собой тонкий однородный материал, спектр записывают в проходящем ИК-излучении, предварительно закрепив образец в специальном держателе. Он представляет собой пластинку с прямоугольным отверстием, к которому прижимается образец, накрываемый сверху магнитной пластинкой с отверстием в центре[26].
ИК-измерения для газообразных веществ требуют гораздо более длинных оптических путей, обычно 10 см при достаточно высокой концентрации. В случае следовых концентраций применяются системы с многократным отражением, обеспечивающие оптический путь порядка нескольких метров при небольших размерах прибора. В этом случае предел обнаружения составляет ниже 1 м. д.[25] Особенностью спектроскопии газообразных образцов является проявление вращательного движения молекул, а также уширение спектральных линий вследствие теплового движения и соударения частиц. С данным видом спектроскопии связан ряд других проблем, например, из-за очень большой длины оптического пути существенную роль начинает играть расходимость светового пучка, из-за чего между центральными и краевыми лучами пучка возникает дополнительная разность хода[29].
ИК-спектроскопия отражения
В традиционной инфракрасной спектроскопии исследуется спектр излучения, прошедшего через образец. Существуют также методы исследования инфракрасного излучения, отражённого от поверхности образца. Они основаны на изучении:
- нарушенного полного внутреннего отражения (НПВО);
- зеркального отражения;
- скользящего отражения;
- диффузного отражения[30].
Существенным преимуществом таких методов является то, что удаётся изучать образцы, непрозрачные для ИК-излучения, а также обходиться без процесса пробоподготовки и проводить анализ непосредственно в полевых условиях. Кроме того, такие анализы не являются разрушающими[30].
Спектроскопия НПВО

Метод основан на отражении пучка на границе раздела двух фаз: фазы кристалла НПВО с относительно высоким показателем преломления и фазы исследуемого образца с более низким показателем преломления. Если пучок излучения падает на плоскость образца под углом падения больше критического, то наблюдается практически полное отражение пучка от поверхности образца. На самом деле излучение на небольшую глубину проникает в фазу образца, где частично поглощается. При последующих попаданиях того же пучка света на образец это явление повторяется, и в результате получается некое подобие спектра поглощения. Наблюдаемые частоты поглощённого излучения будут совпадать с частотами, получаемыми в ИК-спектроскопии пропускания[31].
Для проведения спектроскопии НПВО инфракрасные спектрометры оборудуются специальной приставкой. В ней анализируемое вещество помещается в непосредственный контакт с кристаллом и фиксируется при помощи прижимного устройства. Далее через кристалл под специально подобранным углом подаётся инфракрасное излучение, интенсивность которого фиксируется на выходе из кристалла. Обычно в дисперсионных приборах осуществляется примерно 25 отражений, а в спектрометрах с преобразованием Фурье — около шести[31].
Спектроскопия НПВО позволяет анализировать как обычные жидкие образцы, так и «сложные», например, водные растворы, пасты и гели. Поскольку кристалл НПВО легко извлекается из кюветы, нанесение и удаление образца не представляет особой трудности. Также анализу поддаются порошки и полимеры, которые прижимаются к кристаллу специальным устройством. Существуют специальные кюветы для анализа кожи, которые находят применение при изучении действия косметики и лекарств на кожу человека[31].
Спектроскопия внешнего отражения

Регистрируемым параметром в инфракрасной спектроскопии внешнего отражения является интенсивность отражённого света. Если разделить это значение на интенсивность падающего излучения, получится величина, называемая коэффициентом отражения. График зависимости коэффициента отражения от длины волны (или частоты излучения) содержит ту же информацию, что и классические ИК-спектры пропускания[32].
Спектроскопия зеркального отражения применяется для материалов, нанесённых на отражающие металлические поверхности или поверхности из другого материала, который отражает инфракрасное излучение. Суть метода заключается в том, что пучок излучения из ИК-спектрометра подаётся на изучаемую поверхность, где он проникает сквозь покрытие, отражается от подложки, снова проходит через покрытие и попадает на детектор прибора. Двойное прохождение через материал покрытия приводит к частичному поглощению ИК-излучения, что и даёт спектр поглощения для данного материала. При этом, в отличие от метода НПВО, образец может иметь шероховатую поверхность и не контактирует с кристаллом. Анализу поддаются покрытия толщиной от 1 до 100 мкм[33].
Спектроскопию скользящего отражения применяют для изучения очень тонких слоёв на отражающей поверхности. При подаче излучения под очень большим углом падения оптический путь через слой материала сильно возрастает, что и даёт возможность получать спектры поглощения таких материалов. Если в качестве отражателя выступает вода, то этим методом можно изучать мономолекулярные слои масел, жиров, липидов и т. д. на её поверхности, при этом получая информацию о строении и плёнок. Подобным образом исследуют биологические мембраны в естественных условиях[34].

Диффузное отражение возникает на шероховатой поверхности и не сфокусировано в определённой точке, поэтому для работы с ним используются эллипсоидные зеркала, одно из которых фокусирует ИК-излучение на образце, а второе «собирает» отражённый свет и отправляет его на детектор. Спектроскопия диффузного отражения нашла применение в анализе порошков, а также волокнистых материалов (бумаги, ткани)[35].
Недостатком методов, использующих внешнее отражение, является сложность получаемых спектров. Обычные спектры пропускания несут в себе информацию лишь о коэффициенте экстинкции при той или иной длине волны, в то время как в спектроскопии отражения интенсивность отражённого света зависит также от коэффициента преломления. В дополнение ко всему необходимо учитывать коэффициент поглощения отражающей поверхности. Для преобразования экспериментальных спектров в классические спектры пропускания используют преобразования Крамерса — Кронига[36].
ИК-спектроскопия испускания
Несмотря на то, что большинство инфракрасных спектрометров предназначено для проведения экспериментов с поглощением ИК-излучения, разработаны также методы инфракрасной спектроскопии испускания, в которой регистрируются инфракрасные волны, излучаемые веществом. ИК-спектроскопия испускания демонстрирует большую чувствительность, нежели спектроскопия поглощения, поскольку она имеет нулевой уровень шума. Это означает, что детектором воспринимаются исключительно длины волн, приходящие от изучаемого образца, в то время как в спектроскопии поглощения источник света излучает волны в непрерывном диапазоне длин волн[37].
Для проведения таких экспериментов необходимы специальные условия. Изучаемый образец должен иметь температуру, отличную от температуры спектрометра, иначе будет отсутствовать поток излучения между образцом и детектором. Желательно, чтобы температура образца была выше, поскольку с повышением температуры сильно возрастает интенсивность ИК-излучения от образца. Необходимо также учитывать, что сам спектрометр или нагревательный элемент могут быть источниками мешающего фонового инфракрасного излучения[37]. Избежать обеих проблем позволяет, например, детектор из InSb, охлаждённый до температуры жидкого азота (77 К), и прочие детекторы, охлаждаемые жидким азотом или жидким гелием (4 К), излучением которых можно пренебречь[38].
Типичной областью применения ИК-спектроскопии испускания являются исследования атмосферы: ИК-излучение Земли, проходящее через слой атмосферы, детектируется спутником в направлении надира. При этом излучение Земли имеет спектр чёрного тела, в котором присутствуют полосы поглощения молекул атмосферы. Существуют также методы снятия ИК-спектров испускания жидкостей (например, тонких плёнок расплавов солей), поверхностей и твёрдых тел небольшой толщины (несколько мкм). Важной областью использования спектроскопии испускания является инфракрасная астрономия. Хотя большинство небесных тел дают ИК-спектры поглощения на фоне звёзд или пыли, некоторые объекты, например, кометы, имеют замечательные спектры испускания. В спектрах проявляются горячие испарённые молекулы и продукты их фотолиза. Так, среди обнаруженных данным методом частиц находятся H2O, CO, CO2, C2, CN, CH4, C2H2, C2H, CH3OH, HCN, OCS и СН. Также спектры испускания имеют некоторые планеты-гиганты. Стратосфера Юпитера показывает наличие этана, а в полярном сиянии Юпитера, Сатурна и Урана обнаружено излучение частицы H+
3. Большинство этих спектров было записано криогенными спектрометрами, а некоторые из них сняты Инфракрасной космической лабораторией[39].
Комбинирование с другими методами
С газовой хроматографией
Благодаря своей информативности инфракрасная спектроскопия используется в комбинации с газовой хроматографией. В данном случае и разделение смеси веществ, и запись ИК-спектра проводятся в газообразной фазе. Вещества, выходящие из хроматографической колонки подаются в так называемую «световую трубку» — нагретый позолоченный канал, не позволяющий анализируемым веществам конденсироваться. Вдоль этой же трубки проходит инфракрасное излучение, интенсивность которого детектируется на выходе. Благодаря быстрой регистрации ИК-спектров с преобразованием Фурье становится возможной запись спектров для каждого компонента разделяемой смеси[40].
Необходимо учитывать, что спектры веществ в газовой фазе обычно отличаются от спектров конденсированных веществ. Для получения ИК-спектров соединений, твёрдых или жидких при комнатной температуре, используется техника вымораживания. Выходящие из колонки вещества попадают на пластинку, охлаждаемую жидким азотом, после чего происходит запись спектра прямо с пластинки[40].
Особый интерес представляет комбинация газовой хроматографии, инфракрасной спектроскопии и масс-спектрометрии. Поскольку в ходе ИК-анализа вещество не разрушается, его можно проанализировать и на масс-спектрометре. Такой анализ даёт весьма большой объём аналитической информации, необходимой для идентификации химических соединений[40].
С термогравиметрическим анализом
Термогравиметрический анализ является неоценимым инструментов для характеризации и изучения полимерных материалов. В ходе этого анализа небольшое количество полимера нагревают в инертной атмосфере и следят за уменьшением его массы в зависимости от температуры и времени эксперимента. При этом обычно происходит улетучивание различных пластификаторов и прочих добавок. Если же летучие компоненты неизвестны, их структуру можно устанавливать при помощи ИК-спектроскопии в режиме реального времени. Для этого газообразные вещества инертным газом переносятся в специальную камеру, через которую пропускается инфракрасное излучение[41].
Иногда при нагревании полимеров выделяются не индивидуальные вещества, а их смеси. Несмотря на некоторую информативность ИК-спектров смесей веществ, применяются также и методы их раздельного анализа. Для этого летучие компоненты сорбируют на активированном угле, затем десорбируют и анализируют на газовом хроматографе с детекцией ИК-спектрометром[41].
Двумерная ИК-спектроскопия
Двумерная инфракрасная спектроскопия является относительно новым методом, позволяющим расширить возможности стандартной ИК-спектроскопии. Двумерный ИК-спектр получается при корреляционном анализе динамических флуктуаций сигналов, которые вызываются внешними возмущениями разной природы. Такие спектры находят основное применение при изучении взаимодействий между функциональными группами. Снятие двумерного спектра позволяет упростить перегруженные сигналами одномерные спектры, улучшить разрешение за счёт появления второго измерения и обнаружить корреляции между частями молекулы[42].
В основе двумерной ИК-спектроскопии не может лежать наблюдение обычных колебательных переходов в молекулах, поскольку они происходят весьма быстро (за время порядка пикосекунд), по сравнению, например, со спектроскопией ЯМР, где время релаксации составляет микросекунды, что позволяет записывать двумерные ЯМР-спектры на основе тех же переходов, что и одномерные. В двумерной же ИК-спектроскопии приходится проводить наблюдение других релаксационных процессов, которые индуцируются внешним воздействием. В итоге получается так называемый динамический ИК-спектр, в котором во времени варьируются интенсивности полос, их положение (волновые числа) и направление поглощений (явление дихроизма). В качестве источников внешнего воздействия могут выступать электрические, термические, магнитные, химические, акустические или механические факторы, причём каждый из этих факторов оказывает на систему собственное уникальное воздействие. В результате можно получать двумерные спектры, содержащие различные наборы информации[43].
Применение
Наряду с традиционным использованием в различных областях химии для установления строения и идентификации химических соединений, инфракрасная спектроскопия также нашла применение в других специальных областях.
Исследование памятников искусства
Наряду со спектроскопией комбинационного рассеяния, ИК-спектроскопия находит применение в анализе состава различных предметов искусства. Существенную часть таких приложений составляет анализ неорганических и органических пигментов и красителей. Поскольку инфракрасная спектроскопия позволяет идентифицировать химический состав и строение пигмента, становится возможным сделать ряд косвенных выводов, например, о подлинности или времени реставрации картины. Так, если белый пигмент на полотне периода Ренессанса, согласно данным анализа, представляет собой диоксид титана в форме рутила или анатаза, используемых в изобразительном искусстве с 1923 и 1947 года соответственно, то картина либо подделана, либо недавно подвергалась реставрации[44].
Такие материалы, как лён и хлопок, не удаётся анализировать инфракрасным излучением из-за сильного поглощения молекул воды[45]. Это же относится и к неорганическим пигментам: они имеют невысокие волновые числа и содержат гидроксильные группы в гидратированных кристаллах. Поэтому ИК-спектроскопия имеет более широкое применение в идентификации органических пигментов, связующих и смесей[46]. Особенно важна роль ИК-спектроскопии в исследовании предметов, имеющих флуоресцентное покрытие либо флуоресцентные примеси, поскольку в таких случаях флуоресценция мешает проявлению сигналов КР-спектроскопии[45].
Исследования предметов искусства с использованием ИК-излучения стали проводиться раньше, нежели КР-спектроскопические исследования, и уже собраны достаточно обширные базы ИК-спектров для пигментов, а также синтетических и природных материалов. Подобным анализам также поспособствовало создание портативных приборов, позволяющих анализировать предметы на месте их расположения, например, в музеях[45].
Применение в медицине
Возможность получения информации о присутствии в образце тех или иных функциональных групп позволила использовать инфракрасную спектроскопию в медицинских целях как инструмент изучения биохимии тканей. ИК-спектроскопия, в частности, чувствительна к структуре и концентрации макромолекул (белков, ДНК) и гораздо менее применима для обнаружения небольших молекул, которые находятся в клетках в низкой концентрации. Изменения в ИК-спектрах биологических материалов свидетельствуют о патологиях, связанных с нарушением биохимического состава образца. Например, раковые изменения часто связаны с присутствием нескольких ядер в клетке. Соответственно, инфракрасная спектроскопия показывает диагностические изменения, связанные с усилением поглощения нуклеиновых кислот[47].
Биологические жидкости изучаются в объёме 5—10 мкл методом пропускания через окно из CaF2 или BaF2. При необходимости из получаемых спектров математически вычитается спектр воды. Также воду можно предварительно удалить высушиванием образца и изучать остаток в виде тонкой плёнки, однако в этом случае теряется информация о летучих компонентах образца и о его гидратации. Спектры тканей также получают подобным образом, вырезая и изучая образцы объёмом около 1 мм³. Некоторые ткани, которые невозможно сжать между стёклами (кожа, мышцы), подвергают исследованию методом НПВО[47].
Сбор и интерпретация данных возможны либо классическим методом (изучение интенсивности характеристических полос поглощения по спектрам), либо путём построения пространственных карт интенсивности частот. В последнем случае используется инфракрасный микроскоп, позволяющий снимать спектры последовательно из заданных точек образца, а затем отображать результат в виде трёхмерного графика[47].
Преимуществом такого метода исследования является универсальность прибора: для изучения широкого спектра нарушений в различных тканях не требуется серьёзной перестройки конфигурации или использования специальных детекторов и реагентов[47].
Применение в судебной экспертизе

Основными задачами ИК-спектроскопии в судебной экспертизе являются установление происхождения и марки автомобильных красок, анализ волокон с места преступления, исследование и сравнение типа чернил или тонеров на документах, различение природных и искусственных драгоценных камней, а также анализ пищевых и физиологических образцов[48].
Из-за специфики анализируемых материалов эксперты применяют ряд необычных модификаций инфракрасной спектроскопии. Например, часто используется ячейка с алмазными наковальнями, позволяющая под действием высокого давления расплющить даже очень небольшой образец (порядка 5 мкм) до приемлемой площади, позволяющей запись инфракрасного спектра пропускания. Если образец невозможно переместить, либо если он имеет отражающую поверхность, ИК-спектр записывают в отражении через микроскоп или обычную ячейку. Иногда используется запись спектра из диффузного отражения[49].
См. также
Примечания
Комментарии
- ↑ Нормальными называются колебания, которые не зависят от других колебаний.
- ↑ Это выражение получается из того факта, что каждый из n атомов молекулы имеет 3 степени свободы в трёхмерном пространстве, значит суммарно молекула имеет 3n степеней свободы. Из них три — это поступательные степени свободы, связанные с перемещением целой молекулы в пространстве, ещё две или три — вращательные степени свободы (для линейной молекулы одна из степеней свободы вырождается, поскольку не приводит к изменению энергии). Соответственно, остальные степени свободы являются колебательными.
- ↑ Под режимом накопления подразумевается многократное снятие спектра одного и того же образца и последующее их математическое суммирование, в результате чего интенсивность шума, имеющего случайный характер, снижается в сравнении с единичным спектром, а интенсивность сигналов увеличивается.
- ↑ На самом деле различие в длинах связей изотопных аналогов является лишь кажущимся. Дело в том, что равновесная длина связи рассчитывается как средняя длина между крайними положениями атомов в процессе колебания. Дейтерий колеблется с меньшей амплитудой, чем протий, поэтому средняя длина связи кажется меньшей. Если расчёт проводить для неколеблющейся молекулы, эта разница исчезнет.
Источники
- ↑ 1 2 3 Смит, 1982, с. 9—11.
- ↑ 1 2 ESS, 2010, p. 2938.
- ↑ Larkin, 2011, p. 4.
- ↑ ESS, 2010, p. 2941.
- ↑ ESS, 2010, p. 2943—2944.
- ↑ 1 2 ESS, 2010, p. 2944.
- ↑ 1 2 3 Larkin, 2011, p. 7—8.
- ↑ Larkin, 2011, p. 8—9.
- ↑ Смит, 1982, с. 145.
- ↑ 1 2 Stuart, 2004, p. 8—13.
- ↑ 1 2 3 4 5 6 Larkin, 2011, p. 10—13.
- ↑ Смит, 1982, с. 151—152.
- ↑ Смит, 1982, с. 153.
- ↑ 1 2 Смит, 1982, с. 153—154.
- ↑ 1 2 Larkin, 2011, p. 13—15.
- ↑ Бёккер, 2009, p. 141.
- ↑ 1 2 Бёккер, 2009, с. 156—157.
- ↑ 1 2 Бёккер, 2009, с. 157—160.
- ↑ Stuart, 2004, p. 18—19.
- ↑ Бёккер, 2009, с. 159.
- ↑ 1 2 3 4 5 6 ESS, 2010, p. 1187—1190.
- ↑ Смит, 1982, с. 156—157.
- ↑ 1 2 3 ESS, 2010, p. 1174—1176.
- ↑ 1 2 3 ESS, 2010, p. 2213—2220.
- ↑ 1 2 3 4 ESS, 2010, p. 1210—1217.
- ↑ 1 2 3 Бёккер, 2009, p. 164.
- ↑ Бёккер, 2009, p. 165—166.
- ↑ Бёккер, 2009, p. 166—167.
- ↑ Бёккер, 2009, p. 167—169.
- ↑ 1 2 Бёккер, 2009, с. 169—171.
- ↑ 1 2 3 Бёккер, 2009, с. 171—175.
- ↑ Бёккер, 2009, с. 176.
- ↑ Бёккер, 2009, с. 176—177.
- ↑ Бёккер, 2009, с. 177—178.
- ↑ Бёккер, 2009, с. 179—180.
- ↑ Бёккер, 2009, с. 180—181.
- ↑ 1 2 Bernath, 1996.
- ↑ Bernath2, 2000, p. 183—184.
- ↑ Bernath2, 2000, p. 209—215.
- ↑ 1 2 3 Бёккер, 2009, с. 189—190.
- ↑ 1 2 Бёккер, 2009, с. 190—191.
- ↑ Noda, 1990, p. 550.
- ↑ Noda, 1990, p. 550—551.
- ↑ ESS, 2010, p. 27.
- ↑ 1 2 3 ESS, 2010, p. 23.
- ↑ ESS, 2010, p. 36.
- ↑ 1 2 3 4 ESS, 2010, p. 1494—1497.
- ↑ ESS, 2010, p. 681—692.
- ↑ ESS, 2010, p. 681—682.
Литература
- Бёккер Ю. Спектроскопия = Spektroskopie / Пер. с нем. Л. Н. Казанцевой, под ред. А. А. Пупышева, М. В. Поляковой. — М.: Техносфера, 2009. — 528 с. — ISBN 978-5-94836-220-5.
- Смит А. Прикладная ИК-спектроскопия: основы, техника, аналитическое применение / Пер. с англ. Б. Н. Тарасевича, под ред. А. А. Мальцева. — М.: Мир, 1982. — 328 с.
- Bernath P. F. Infrared fourier transform emission spectroscopy (англ.) // Chem. Soc. Rev. — 1996. — Vol. 25. — P. 111—115. — doi:10.1039/CS9962500111.
- Bernath P. F. Infrared emission spectroscopy (англ.) // Annu. Rep. Prog. Chem., Sect. C: Phys. Chem. — 2000. — Vol. 96. — P. 177—224. — doi:10.1039/B001200I. Архивировано 2 апреля 2015 года.
- Encyclopedia of Spectroscopy and Spectrometry / Lindon J. — 2nd Ed. — Academic Press, 2010. — 3312 p.
- Larkin P. J. Infrared and raman spectroscopy: principles and spectral interpretation. — Elsevier, 2011. — 230 p. — ISBN 978-0-12-386984-5.
- Noda I. Two-Dimensional Infrared (2D IR) Spectroscopy: Theory and Applications (англ.) // Applied Spectroscopy. — 1990. — Vol. 44, no. 4. — P. 550—561.
- Stuart B. H. Infrared Spectroscopy: Fundamentals and Applications. — Wiley, 2004. — 242 p.
Ссылки
Базы данных ИК-спектров
- Spectral Database for Organic Compounds SDBS. Дата обращения: 21 октября 2014.
- NIST Chemistry WebBook. Дата обращения: 21 октября 2014.
Учебные материалы
- Royal Society of Chemistry. Infra-Red Spectroscopy (IR) — видео. Дата обращения: 22 октября 2014.