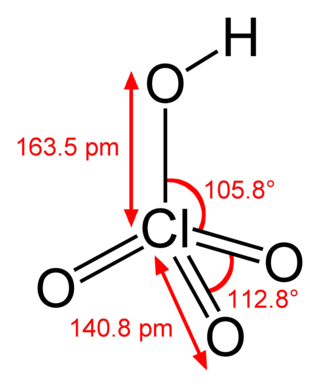
Кислоты
Кисло́ты — химические соединения, способные отдавать катион водорода (кислоты Брёнстеда), либо соединения, способные принимать электронную пару с образованием ковалентной связи (кислоты Льюиса)[1].
В быту и технике под кислотами обычно подразумеваются кислоты Брёнстеда, образующие в водных растворах избыток ионов гидроксония H3O+. Присутствие этих ионов обуславливает кислый вкус растворов кислот, способность менять окраску индикаторов и, в высоких концентрациях, вызывать раздражающее действие. Подвижные атомы водорода кислот способны замещаться на атомы металлов с образованием солей, содержащих катионы металлов и анионы кислотного остатка.
Определение кислоты
История развития представлений о кислотах

Кислоты как класс химических соединений, обладающих рядом близких свойств, известны с древнейших времён.
В 1778 году французский химик Антуан Лавуазье предположил, что кислотные свойства обусловлены наличием в их составе кислорода. Эта гипотеза оказалась несостоятельной, так как многие кислоты не имеют в своём составе кислорода, в то время как многие кислородсодержащие соединения не проявляют кислотных свойств. Тем не менее, именно эта гипотеза дала название кислороду как химическому элементу[2]. В 1833 году немецкий химик Юстус Либих определил кислоту как водородсодержащее соединение, в котором водород может быть замещён на металл[2].
Первую попытку создать общую теорию кислот и оснований предпринял шведский физикохимик Сванте Аррениус. В его теории, сформулированной в 1887 году, кислота определялась как соединение, диссоциирующее в водном растворе с образованием ионов водорода H+[2]. Теория Аррениуса быстро показала свою ограниченность. Во-первых, было выяснено, что невозможно представить существование несольватированного катиона Н+ в растворе; во-вторых, теория Аррениуса не учитывала влияние растворителя на кислотно-основные равновесия; наконец, теория оказалась неприменима к неводным системам[3].
Согласно сольвентной теории Франклина, созданной в 1924 году, кислотой называлось вещество, при растворении увеличивавшее число тех же катионов, которые образуются при диссоциации растворителя. Данная теория сыграла важную роль в исследовании неводных растворов кислот. Химическая теория кислот и оснований формировалась в работах А. Ганча (1917—1927). По Ганчу, кислотами называются соединения водорода, в которых последний может быть замещён на металл или неметаллический радикал с образованием соли[2].
В 1923 году появились теория кислот и оснований Брёнстеда-Лоури и теория кислот Льюиса, широко применяемые по настоящее время[4].
Кислота в теории Брёнстеда-Лоури

В протонной теории кислот и оснований, выдвинутой в 1923 г. независимо и почти одновременно датским учёным Й. Брёнстедом и английским учёным Т.-М. Лоури (Лаури), кислотой называется соединение или молекулярный ион, способные отдавать протон другому химическому соединению — основанию[5]. Согласно теории Брёнстеда-Лоури, в химическом взаимодействии кислот и оснований всегда принимают участие кислота — донор протона (кислота Брёнстеда) и сопряженное с ней основание — любое соединение, способное присоединять протон (основание Брёнстеда). Поскольку основание может быть рассмотрено как продукт отщепления протона от кислоты, электрический заряд сопряженной кислоты всегда на единицу меньше, чем заряд сопряженного с ней основания. Так, например, кислоте HCl соответствует сопряженное с ней основание — хлорид-ион Cl−[2][6].
Кислота в теории Льюиса

Согласно электронной теории, предложенной в 1923 году американским физикохимиком Г. Н. Льюисом, кислота — это вещество, принимающее электронную пару и образующее связь с основанием Льюиса за счёт этой пары электронов[7]. Кислотами в теории Льюиса могут быть молекулы с незаполненной восьмиэлектронной оболочкой (BF3, SO3), катионы металлов-комплексообразователей (Fe2+, Zn2+), галогениды переходных металлов (TiCl4), молекулы с полярными двойными связями (SO2), карбокатионы. По сравнению с теорией Брёнстеда-Лоури, теория Льюиса является более общей и охватывает более широкий круг кислот[3][4].
Ключевым свойством, определяющим способность взаимодействия кислоты Льюиса с основанием Льюиса, является энергетическое соответствие между низшей свободной молекулярной орбиталью, принимающей электронную пару, и высшей занятой молекулярной орбиталью, с которой эта электронная пара уходит. Эта способность была учтена в рамках принципа жёстких и мягких кислот и оснований Пирсона (принцип ЖМКО). Данный принцип устанавливает, что мягкие кислоты наиболее склонны взаимодействовать с мягкими основаниями и жёсткие кислоты — с жёсткими основаниями. При этом под жёсткими кислотами следует понимать кислоты Льюиса, обладающие большим положительным зарядом, большой электроотрицательностью и низкой поляризуемостью. Напротив, мягкие кислоты обладают малым положительным зарядом, низкой электроотрицательностью и высокой поляризуемостью. Поскольку данные свойства изменяются плавно, ряд кислот Льюиса занимает промежуточное положение между жёсткими и мягкими[4]. Принцип ЖМКО не имеет количественного критерия оценки силы кислот, поэтому он не может быть применён для аналитических расчётов[3].
| Жёсткие кислоты | Промежуточные кислоты | Мягкие кислоты |
|---|---|---|
| H+, Li+, Na+, K+, Mg2+, Ca2+, Al3+, Cr3+, Fe3+, BF3, B(OR)3, AlR3, AlCl3, SO3, RCO+, CO2, RSO2+ | Cu2+, Fe2+, Zn2+, SO2, R3C+, C6H5+, NO+ | Ag+, Cu+, Hg2+, RS+, I+, Br+, Pb2+, BH3, карбены |
| Жёсткие основания | Промежуточные основания | Мягкие основания |
| OH−, RO−, F−, Cl−, RCOO−, NO3−, NH3, RNH2, H2O, ROH, SO42−, CO32−, R2O, NR2−, NH2− | Br−, C6H5NH2, NO2−, C5H5N | RS−, RSH, I−, H−, R3C−, алкены, C6H6, R3P, (RO)3P |
Кислота в общей теории Усановича
В 1939 году М. И. Усанович сформулировал общую теорию кислот и оснований, согласно которой кислотой является частица, которая может отдавать катионы, в том числе протон, или присоединять анионы, в том числе электрон. Таким образом, понятие кислоты, по Усановичу, включает как кислоты Брёнстеда, так и кислоты Льюиса, а также окислители[3]. Кроме того, само понятие кислотности, как и основности, в общей теории Усановича рассматривается не как функция вещества как такового, а как роль, которую оно играет в зависимости от партнёра по реакции[9].
Классификация кислот

Кроме подразделения на кислоты Льюиса и кислоты Брёнстеда, последние принято классифицировать по различным формальным признакам:
- бескислородные (HCl, H2S, HCN) — водные растворы галогенидов, псевдогалогенидов и низших халькогенидов;
- кислородсодержащие (HNO3, H2SO4, C6H5COOH). Неорганические кислородсодержащие кислоты являются кислотными гидроксидами.
- По силе
- Сильные — диссоциируют практически полностью, константы диссоциации больше 1⋅10−3 (HNO3);
- Слабые — константа диссоциации меньше 1⋅10−3 (уксусная кислота Ka= 1,7⋅10−5).
- По устойчивости
- По принадлежности к классам химических соединений
- Неорганические (HBr, H2SO4);
- Органические (HCOOH, CH3COOH, C3H2O2);
- По летучести
- По растворимости в воде
- Растворимые (H2SO4);
- Нерастворимые (H2SiO3);
- По содержанию атомов металлов
Номенклатура кислот
Номенклатура неорганических кислот
Названия кислородсодержащих кислот состоят из двух частей: собственного названия кислоты, выраженного прилагательным, и группового слова кислота (серная кислота, фосфорная кислота). Собственное название кислоты образуется от русского названия кислотообразующего элемента путём добавления различных суффиксов:
- -н-, -ов-, -ев- (если элемент находится в единственной или высшей степени окисления);
- промежуточная степень окисления +5 обозначается суффиксом -новат- (хлорноватая кислота HClO3, бромноватая кислота HBrO3, иодноватая кислота HIO3);
- промежуточные степени окисления +3 и +4 обозначаются суффиксом -(ов)ист- (мышьяковистая кислота HAsO2, хлористая кислота HClO2);
- степень окисления +1 обозначается суффиксом -новатист- (азотноватистая кислота H2N2O2, хлорноватистая кислота HClO).
Если кислотообразующий элемент в двух кислотах находится в одной и той же степени окисления, но кислоты отличаются по «содержанию воды», то для кислоты с меньшим содержанием кислорода к названию добавляют приставку мета-, а для кислоты с большим содержанием кислорода — приставку орто-, например, метафосфорная кислота HPO3 и ортофосфорная кислота H3PO4.
Кислородсодержащие кислоты с несколькими кислотообразующими элементами называются изополикислотами. Их обычно называют традиционными названиями (дифосфорная кислота H4P2O7, дисерная кислота H2S2O7).
Кислоты, в которых атомы кислорода заменены на атомы серы, называются тиокислотами и имеют соответствующую приставку тио- (тиофосфорная кислота H3PO3S). Если гидроксильные группы кислоты или атомы кислорода замещены на атомы галогенов или аминогруппу, то к названию также добавляется соответствующая приставка (амидофосфорная кислота H2PO3NH2), а замещённые серные кислоты по традиции называют сульфоновыми (хлорсульфоновая кислота ClSO3H).
Кислоты с пероксидным мостиком –O–O– относятся к пероксокислотам и имеют приставку пероксо- (пероксомоносерная кислота H2SO5) либо над- (надсерная кислота)[11].
В систематических названиях кислот к корню латинского названия кислотообразующего элемента добавляют суффикс -ат, а названия остальных элементов или их групп в анионе обозначаются приставками. В скобках указывают степень окисления кислотообразующего элемента, если она имеет целочисленное значение. В противном случае в название включают и число атомов водорода: HClO4 — тетраоксохлорат(VII) водорода (хлорная кислота), HAuCl4 — тетрахлороаурат(III) водорода (золотохлористоводородная кислота), H[Sb(OH)6] — гексагидроксостибат(V) водорода и т. д.[12]
Номенклатура органических кислот
Традиционно для простейших карбоновых кислот наиболее распространены тривиальные названия, некоторые из которых образовались ещё в XVII веке (уксусная кислота, масляная кислота, адипиновая кислота, фталевая кислота). Высшие карбоновые кислоты с чётным числом атомов углерода также имеют тривиальные названия, которые, однако, так сходны, что их употребление может вызывать путаницу (каприловая кислота, каприновая кислота).
Систематические названия карбоновых кислот образуются путём добавления окончания -овая кислота к названию соответствующего кислоте алкана (гексановая кислота, пентакозановая кислота). В случае дикарбоновых кислот используется окончание -диовая кислота (декандиовая кислота). Иногда название более удобно образовывать при помощи окончания -карбоновая кислота, которое означает замену одного атома водорода в соединении на карбоксильную группу. Такой подход применяется в тех случаях, когда карбоксильная группа присоединена к циклической системе (циклопропанкарбоновая кислота).
Если в карбоновой кислоте содержится пероксидный мостик, то к названию таких кислот добавляются приставки перокси-, пер- или над- (надуксусная кислота, пероксибензойная кислота).
Для обозначения серосодержащих органических кислот используют окончания -сульфоновая кислота (RSO3H), -сульфиновая кислота (RSO2H), -сульфеновая кислота (RSOH), аналогичным образом добавляя их к названию родоначального алкана RH[13].
| Формула | Название по ИЮПАК | Тривиальное название | Происхождение тривиального названия |
|---|---|---|---|
| НСООН | метановая кислота | муравьиная кислота | лат. formica — муравьи |
| СН3-СООН | этановая кислота | уксусная кислота | лат. acetum — уксус |
| СН3-СН2-СООН | пропановая кислота | пропионовая кислота | др.-греч. proto + pion — первый + жир |
| СН3-(СН2)2-СООН | бутановая кислота | масляная кислота | лат. butyrum — масло |
| СН3-(СН2)3-СООН | пентановая кислота | валериановая кислота | лат. Valeriána — валериана |
| СН3-(СН2)4-СООН | гексановая кислота | капроновая кислота | лат. caper — коза |
| СН3-(СН2)5-СООН | гептановая кислота | энантовая кислота | др.-греч. oenanthe — цветок винограда |
| СН3-(СН2)6-СООН | октановая кислота | каприловая кислота | лат. caper — коза |
| СН3-(СН2)7-СООН | нонановая кислота | пеларгоновая кислота | лат. Pelargonium — пеларгония |
| СН3-(СН2)8-СООН | декановая кислота | каприновая кислота | лат. caper — коза |
| СН3-(СН2)9-СООН | ундекановая кислота | ундециловая кислота | |
| СН3-(СН2)10-СООН | додекановая кислота | лауриновая кислота | лат. Laurus — лавр |
| СН3-(СН2)11-СООН | тридекановая кислота | тридециловая кислота | |
| СН3-(СН2)12-СООН | тетрадекановая кислота | миристиновая кислота | лат. Myristica — мускатный орех, др.-греч. mύρων — оливковое масло |
| СН3-(СН2)13-СООН | пентадекановая кислота | пентадециловая кислота | |
| СН3-(СН2)14-СООН | гексадекановая кислота | пальмитиновая кислота | лат. palma — пальмовое дерево |
| СН3-(СН2)15-СООН | гептадекановая кислота | маргариновая кислота | др.-греч. margaron — жемчуг |
| СН3-(СН2)16-СООН | октадекановая кислота | стеариновая кислота | др.-греч. stear — сало |
| СН3-(СН2)17-СООН | нонадекановая кислота | нонадециловая кислота | |
| С6Н5-СООН | бензолкарбоновая кислота | бензойная кислота | |
| СН2=СН-СООН | пропеновая кислота | акриловая кислота | лат. acer + olere — острый запах |
| СН≡С-СООН | пропиновая кислота | пропиоловая кислота | |
| СН3-С(СН3)2-СООН | 2,2-диметилпропановая | пивалиновая кислота | сокр. от пинаколин + валериановая кислота[К 1] |
Диссоциация и сила кислот
Количественное описание силы кислот
Теория кислот и оснований Брёнстеда, рассматривающая кислоту, как частицу, способную отдавать протон, даёт возможность количественно оценить эту способность кислоты — её силу. Сила кислот описывается при помощи константы равновесия реакции диссоциации кислоты в водном растворе, называемой также константой кислотности Ka. Чем больше значение Ka, тем больше способность кислоты отдавать протон и тем выше её сила. Также константа кислотности выражается в виде более удобной величины pKa — отрицательного логарифма величины Ka. Например, уравнение диссоциации и константу кислотности плавиковой кислоты можно записать следующим образом[16][17]:

![{\displaystyle K_{\mathrm {a} }={\frac {[{\mathsf {H_{3}O^{+}}}]\cdot [{\mathsf {F^{-}}}]}{[{\mathsf {HF}}]}}=6{,}61\cdot 10^{-4};}](https://wikimedia.org/api/rest_v1/media/math/render/svg/b89f809ea632b8eabdbe7dfe29ecb4c6c84a8025)
Для многоосновных кислот используют несколько значений констант диссоциации Ka1, Ka2 и т. д., соответствующих каждой ступени диссоциации. Например, фосфорная кислота диссоциирует по трём ступеням[17]:

![{\displaystyle {\mathsf {H_{3}PO_{4}+H_{2}O}}\rightleftharpoons {\mathsf {H_{2}PO_{4}^{-}+H_{3}O^{+}}};K_{\mathrm {a1} }={\frac {[{\mathsf {H_{2}PO_{4}^{-}}}]\cdot [{\mathsf {H_{3}O^{+}}}]}{[{\mathsf {H_{3}PO_{4}}}]}}=7{,}52\cdot 10^{-3};}](https://wikimedia.org/api/rest_v1/media/math/render/svg/3933d55cf578bcf29c7ea10c64304e16dcacf270)
![{\displaystyle {\mathsf {H_{2}PO_{4}^{-}+H_{2}O}}\rightleftharpoons {\mathsf {HPO_{4}^{2-}+H_{3}O^{+}}};K_{\mathrm {a2} }={\frac {[{\mathsf {HPO_{4}^{2-}}}]\cdot [{\mathsf {H_{3}O^{+}}}]}{[{\mathsf {H_{2}PO_{4}^{-}}}]}}=6{,}31\cdot 10^{-8};}](https://wikimedia.org/api/rest_v1/media/math/render/svg/7daa1e198ee16fcc7386f32cac56765547858eb6)
![{\displaystyle {\mathsf {HPO_{4}^{2-}+H_{2}O}}\rightleftharpoons {\mathsf {PO_{4}^{3-}+H_{3}O^{+}}};K_{\mathrm {a3} }={\frac {[{\mathsf {PO_{4}^{3-}}}]\cdot [{\mathsf {H_{3}O^{+}}}]}{[{\mathsf {HPO_{4}^{2-}}}]}}=1{,}26\cdot 10^{-12}.}](https://wikimedia.org/api/rest_v1/media/math/render/svg/0f607d39b742ab61cc22e9fe3c4ae31b7bea7a5a)
| Кислота | Значение (m — n) | Ka |
|---|---|---|
| HClO | 0 | 10−8 |
| H3AsO3 | 0 | 10−10 |
| Н2SО3 | 1 | 10−2 |
| Н3РО4 | 1 | 10−2 |
| HNO3 | 2 | 101 |
| H2SO4 | 2 | 103 |
| HClO4 | 3 | 1010 |
Кислоты принято условно подразделять по их силе на очень сильные (pKa < 0), сильные (0 < pKa < 4,5), средней силы (4,5 < pKa < 9), слабые (9 < pKa < 14), очень слабые (pKa > 14)[2].
Для приблизительной оценки силы кислот применяют эмпирические правила Полинга. Так, для неорганических кислородсодержащих кислот вида HnXOm известно эмпирическое правило, по которому значение первой константы связано со значением (m — n). Если A=(m — n) = 0, то кислота очень слабая, при 1 — слабая, при 2 — сильная, и, наконец, при 3 кислота очень сильная[18]. Кроме того, если такую кислоту записать в виде (HO)aXOb, выделив отдельно атомы кислорода, входящие в состав гидроксильных групп (заметим, что b = m ― n), то величину константы диссоциации по первой ступени можно оценить по уравнению:
то есть величина первой константы диссоциации определяется, в основном, числом негидроксильных атомов кислорода b. Эту зависимость связывают с отрицательным индуктивным влиянием этих атомов кислорода на связи O–H, за счёт которого облегчается отщепление протона от молекулы кислоты[19]. Л. Полингу также приписывают эмпирическое правило, связанное с кислотностью многоосновных кислот, которое говорит, что последовательные константы диссоциации многоосновных кислот pKa1, pKa2, pKa3 находятся в отношении 1 : 10−5 : 10−10. Уменьшение констант кислотности связано с увеличением заряда образующегося аниона[19]. Рассчитанные по правилам Полинга значения pKa отличаются от экспериментальных всего на ±1[20]. Другой характеристикой силы кислоты может служить степень диссоциации α — отношение количества диссоциированных на ионы молекул кислоты к их исходному количеству в растворе. Степень диссоциации выражается в процентах либо в виде безразмерной величины от 0 до 1[21]:

![{\displaystyle \alpha ={\frac {C_{\text{dis}}}{C_{0}}}={\frac {[{\mathsf {X^{-}}}]}{[{\mathsf {HX}}]+[{\mathsf {X^{-}}}]}}.}](https://wikimedia.org/api/rest_v1/media/math/render/svg/a9b3da9542f921316247e2eb4ddbd3921805f767)
| Формула кислоты | pKa | Формула кислоты | pKa |
|---|---|---|---|
| HClO4 | -5 ± 0,5 | H2PO4- | 7,20 |
| H2SO4 | -2,8 ± 0,5 | HClO | 7,25 |
| H3O+ | -1,74 | H3BO3 | 9,24 |
| HNO3 | -1,32 | NH4+ | 9,25 |
| (COOH)2 | 1,26 | HCN | 9,22 |
| H2SO3 | 1,92 | HCO3- | 10,33 |
| HSO4- | 1,96 | H2O2 | 11,62 |
| H3PO4 | 2,12 | HPO42- | 12,32 |
| HF | 3,14 | H2O | 15,74 |
| HNO2 | 3,35 | NH3 (ж.) | 33 |
| CH3COOH | 4,76 | H2 | 38,6 |
| H2S | 7,05 | СH4 | ~58 |
Влияние растворителя
Нивелирование и дифференцирование кислот
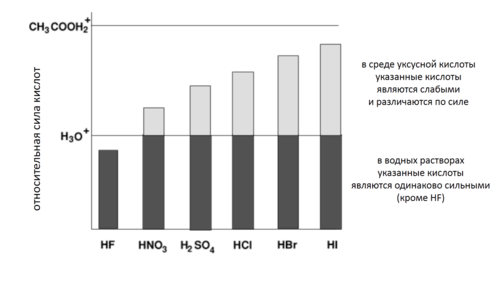
На кислотно-основное равновесие значительное влияние оказывает растворитель. В частности, для водных растворов было обнаружено, что все кислоты с константами кислотности pKa < 0 имеют одинаковые свойства (например, pH растворов). Объясняется это тем, что для таких кислот равновесие практически нацело смещено в сторону образования иона гидроксония H3O+, который является единственной кислотой в растворе. По сути, ион гидроксония представляет собой самую сильную кислоту в водных растворах, поскольку все более сильные кислоты нацело превращаются в него и таким образом выравниваются по своей силе. Например, все кислоты с pKa < 0 (HCl, HBr, H2SO4) нацело диссоциированы в водных растворах.
Аналогичная ситуация наблюдается и в других растворителях: если pKa протонной кислоты в системе «кислота-растворитель» превосходит кислотность протонированного растворителя (его сопряженной кислоты), то происходит полный перенос протонов от кислоты к растворителю и единственной и наиболее сильной кислотой в растворе становятся катионы протонированного растворителя; кислотность раствора при этом определяется кислотностью сопряженной кислоты растворителя. Данное явление получило название нивелирующего эффекта растворителя[22]. Аналогичным образом, и очень слабые кислоты с pKa > 14 в водных растворах выравниваются по силе[23][24].
Кислотность протонированной уксусной кислоты CH3CO2+H2 значительно выше, чем кислотность протонированной воды H3O+, поэтому нивелирующий эффект в уксуснокислых растворах наблюдается при значительно более низких pKa кислот.
Кислоты с pKa от 0 до 14 в воде диссоциированы не полностью: их кислотные свойства в растворе зависят от значения pKa. Например, монохлоруксусная и дихлоруксусная кислоты с pKa 2,86 и 1,26 соответственно сильно отличаются по степени диссоциации (в 0,1 М растворе первая диссоциирует на 11 %, а вторая — на 52 %)[25]. В этом случае говорят о дифференцирующем эффекте растворителя. Интервал pKa, в котором кислоты дифференцированы по силе, равен показателю константы автопротолиза растворителя. Для разных растворителей этот интервал различен (14 для воды, 19 для этанола, 33 для аммиака и т. д.), соответственно, и набор дифференцированных и нивелированных кислот для них разный[26].
Если растворитель обладает основными свойствами, то в нём все кислоты становятся более сильными и большее число кислот нивелируется по силе (например, в аммиаке уксусная кислота диссоциирует нацело, хотя в водных растворах она имеет среднюю силу). Напротив, если основные свойства растворителя понижаются, то сильные кислоты могут стать слабыми, а число нивелированных кислот понижается[26]. Например, уксусная кислота служит нивелирующим растворителем для хлорной кислоты HClO4, диссоциирующей в ней нацело, и для хлороводородной кислоты HCl, и для серной кислоты H2SO4, являющихся в уксусной кислоте слабыми[27].
Влияние диэлектрической проницаемости
На диссоциацию кислот сильное действие оказывает диэлектрическая проницаемость растворителя. Это влияние особенно заметно при сравнении двух кислот разного зарядового типа. При диссоциации нейтральной кислоты, например, фенола C6H5OH, в растворе образуются два иона. Положительно заряженные кислоты, например, ион триэтиламмония (C2H5)NH+, диссоциируют с образованием одного положительно заряженного иона. Таким образом, в первом случае после реакции число ионов увеличивается, а во втором случае это число не изменяется[28].

Следовательно, переход от растворителя с высокой диэлектрической проницаемостью (более полярных) к растворителям с меньшей диэлектрической проницаемостью (менее полярным) должен сильно уменьшать силу нейтральных кислот и сравнительно мало влиять на заряженные кислоты. Так, в воде фенол в 5 раз сильнее иона триэтиламмония, однако, в метаноле фенол в 2500 раз слабее этого иона[28].

Влияние специфической сольватации анионов
Растворитель может сильно увеличивать кислотность веществ, специфически стабилизируя анионы, образующиеся в результате диссоциации. Так, кислотность веществ в воде, выражающаяся в их депротонировании, определяется как диэлектрической проницаемостью воды (электростатическая сольватация), так и ее способностью образовывать водородные связи (специфическая сольватация), в то время как, например, депротонирование веществ в нитрометане, не склонном к образованию водородных связей, определяется только их способностью к электростатической сольватации[29].
Влияние строения кислот на их силу
Существует несколько факторов, которые определяют относительную силу органических и неорганических кислот и которые связаны со строением той или иной кислоты. Часто несколько факторов действуют одновременно, поэтому трудно предсказать их суммарное влияние. Среди наиболее значимых можно выделить следующие факторы[30].
- Индуктивный эффект (эффект поля) заместителей при кислотной группировке. Электроотрицательные заместители, обладающие отрицательным индуктивным эффектом (–I-эффектом), повышают силу кислот. Например, нитроуксусная кислота O2NCH2COOH почти в 1000 раз более сильная, чем уксусная кислота CH3COOH (pKa равны 1,68 и 4,76 соответственно). Причиной этого является стабилизирующее действие, оказываемое этими заместителями на отрицательный заряд кислотного аниона, образующегося при отщеплении протона. Даже в тех случаях, когда кислота заряжена положительно, а сопряжённое основание не имеет заряда, электроотрицательные группы повышают её кислотность, поскольку они дестабилизируют молекулу кислоты, повышая величину положительного заряда. Напротив, донорные заместители, обладающие +I-эффектом, понижают силу кислот. Индуктивный эффект снижается при увеличении расстояния между кислотным центром и влияющим на него заместителем[4][30].

- Мезомерный эффект (резонансный эффект). Если в анионе, образующемся при диссоциации кислоты, отрицательный заряд распределён между несколькими атомами за счёт явления резонанса, то такой анион стабилизируется и кислотность соединения возрастает. По этой причине карбоновые кислоты — более сильные кислоты, чем соответствующие им спирты (отрицательный заряд карбоксилат-ионов распределён между двумя атомами кислорода). Подобный эффект реализуется также в фенолах, где в стабилизации отрицательного заряда сопряжённого основания принимает участие ароматическая система[4][30].
- Корреляция с расположением атомов в периодической системе. Чем выше электроотрицательность элемента, с которым связан кислотный протон, тем выше сила кислоты. По этой причине кислотность увеличивается при движении вдоль периода периодической системы слева направо. Также кислотность увеличивается при переходе по группе сверху вниз, что связано с увеличением радиуса кислотообразующего атома и, следовательно, меньшей плотностью заряда на нём, что в итоге приводит к более лёгкой диссоциации[30]:


- Закономерности в изменении силы кислот Льюиса также связаны с расположением центрального элемента в периодической системе химических элементов. Так, более сильными кислотами Льюиса оказываются те молекулы, которым для завершения внешнего электронного слоя недостаёт только одной электронной пары. По этой причине, например, хлорид галлия(III) GaCl3 является более сильной кислотой, чем хлорид цинка ZnCl2. Также, при прочих равных условиях, менее сильной является такая кислота MXn, центральный атом которой больше по размеру, что связано с ослаблением взаимодействия между положительно заряженным ядром и электронной парой основания, размещаемой на вакантной орбитали кислоты при кислотно-основной взаимодействии[30].
- Статические эффекты. В случае симметричных двухосновных кислот константа диссоциации по первой ступени в два раза больше ожидаемой, поскольку в кислоте присутствует два протона, способных к ионизации. В то же время константа диссоциации по второй ступени в два раза меньше ожидаемой, поскольку дианион дикарбоновой кислоты может присоединять протон по двум эквивалентным положением. Таким образом, для дикарбоновых кислот с удалёнными друг от друга кислотными группами соотношение Ka1/Ka2 приблизительно равно 4[30].
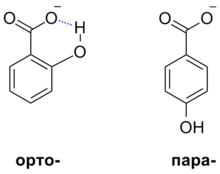
- Водородная связь. Внутримолекулярные водородные связи могут оказывать стабилизирующее влияние на образующиеся анионы и таким образом повышать силу соответствующих кислот. Так, pKa орто-гидроксибензойной кислоты составляет 2,98, а для пара-гидроксибензойной кислоты — лишь 4,58, поскольку для первого соединения возможна реализация внутримолекулярной водородной связи, а для второго нет[30].
- Пространственные эффекты. Для кислот Брёнстеда пространственные затруднения в кислотно-основных реакциях редки, поскольку протон достаточно мал и подвижен. Пространственные эффекты гораздо более выражены для кислот и оснований Льюиса. Например, при использовании объёмной кислоты Льюиса три(трет-бутил)борана t-Bu3B сила взаимодействия определяется не силой основания, а пространственной доступностью его электронной пары[30].
- Гибридизация. Алкины и HCN являются сравнительно более сильными кислотами, чем аналоги с двойной связью и одинарной связью, поскольку анионы при sp-гибридном атоме более устойчивы, чем при sp2- или sp3-гибридном атоме[30].
Функция кислотности Гаммета и суперкислоты
Количественную оценку кислотно-основных свойств очень сильных или концентрированных кислот невозможно провести с использованием шкалы pH, поскольку для водных растворов эта шкала ограничена снизу значением pH = 0, которому соответствует 5%-ый раствор серной кислоты H2SO4. В то же время такая необходимость возникает. Для решения данной задачи в 1932 году Л. Гамметом и А. Дейрупом была предложена функция кислотности Гаммета H0. Их подход заключался в изучении кислотно-основного равновесия очень сильных кислот в присутствии менее сильного основания, чем вода, и измерении соотношения концентраций протонированной и непротонированной форм этого основания методом электронной спектроскопии. Это дало возможность продлить шкалу кислотности в отрицательную области, благодаря чему стала возможной оценка кислотности концентрированных растворов неорганических кислот[31].
Введённая Гамметом шкала кислотности широко применяется для оценки силы суперкислот — сред с кислотностью выше, чем кислотность 100%-ой серной кислоты[32], функция кислотности Гаммета для которой составляет H0 = −12. Среди индивидуальных неорганических соединений сильными кислотами являются хлорная кислота HClO4 (H0 = −13), хлорсульфоновая кислота ClSO3H (H0 = −13,8) и фторсульфоновая кислота FSO3H (H0 = −15,1). Самой сильной из известных органических кислот является трифторметансульфокислота CF3SO3H (H0 = −14,1)[31].
К суперкислотам относятся также смеси кислот Брёнстеда и кислот Льюиса, например, смесь HF и фторида сурьмы(V) SbF5 в разных соотношениях (H0 < −30 при соотношении 1:1). Известным примером суперкислоты является олеум, где роль кислоты Льюиса выполняет SO3, который при реакции с серной кислотой даёт суперкислоту H2S2O7 (H0 = −14,5)[31].
Химические свойства кислот


- Взаимодействие с основными оксидами с образованием соли и воды:

- Взаимодействие с амфотерными оксидами с образованием соли и воды:

- Взаимодействие со щелочами с образованием соли и воды (реакция нейтрализации):

- Взаимодействие с нерастворимыми основаниями с образованием соли и воды, если используемая кислота растворима[33]:


- Сильные кислоты вытесняют более слабые из их солей:


(в данном случае образуется неустойчивая угольная кислота H2CO3 , которая сразу же распадается на воду и углекислый газ)
- Металлы, стоящие в ряду активности до водорода, вытесняют его из раствора кислоты (кроме азотной кислоты HNO3 любой концентрации, концентрированной серной кислоты H2SO4 и других концентрированных кислот, проявляющих ярко-выраженные оксилительные свойства), если образующаяся соль растворима:

- С кислотами — сильными окислителями, например, азотной кислотой и концентрированной серной кислотами реакция идёт с образованием продуктов восстановления аниона кислоты, а не протона:

- Для кислот характерна реакция этерификации (взаимодействие со спиртами с образованием сложного эфира и воды):

Например:

- В неполярном растворителе (например, бензоле) молекулы кислоты способны превращаться в димеры:

Получение кислот
Кислоты получают:
- Путём взаимодействия неметаллов с водородом:


- Путём взаимодействия серной кислоты с твёрдыми солями:
Нелетучая серная кислота при нагревании способна вытеснять летучие кислоты из их солей (таким путём нельзя получить бромоводород и иодоводород из-за их окисления концентрированной серной кислотой)[34].


- Путём взаимодействия кислотных оксидов с водой:

См. также
Комментарии
- ↑ Пивалиновая кислота может быть получена окислением пинаколина и изомерна валериановой кислоте.
Примечания
- ↑ IUPAC Gold Book — Acid. Дата обращения: 15 апреля 2013. Архивировано 17 апреля 2013 года.
- ↑ 1 2 3 4 5 6 7 Химическая энциклопедия / Под ред. И. Л. Кнунянца. — М.: Большая Российская энциклопедия, 1992. — Т. 2. — С. 393—395. — ISBN 5-85270-039-8.
- ↑ 1 2 3 4 Золотов Ю. А., Дорохова Е. Н., Фадеева В. И. и др. Основы аналитической химии. Книга 1. Общие вопросы. Методы разделения / Под ред. Ю. А. Золотова. — 2-е изд., перераб. и доп. — М.: Высшая школа, 1999. — С. 118. — ISBN 5-06-003558-1.
- ↑ 1 2 3 4 5 6 Москва В. В. Понятие кислоты и основания в органической химии // Соросовский образовательный журнал. — 1996. — № 12. — С. 33—40. Архивировано 5 октября 2013 года.
- ↑ IUPAC Gold Book — Brønsted acid. Дата обращения: 15 апреля 2013. Архивировано 17 апреля 2013 года.
- ↑ IUPAC Gold Book — conjugate acid–base pair. Дата обращения: 15 апреля 2013. Архивировано 17 апреля 2013 года.
- ↑ IUPAC Gold Book — Lewis acid. Дата обращения: 15 апреля 2013. Архивировано 17 апреля 2013 года.
- ↑ Золотов и др., 1999, с. 152.
- ↑ Кусаинова К. М. Нет ни кислот, ни оснований! Об одной полузабытой теории и её творце // Химия и жизнь. — 2004. — № 6. — С. 40—44. Архивировано 27 июня 2013 года.
- ↑ 1 2 Рудзитис Г. Е., Фельдман Ф. Г. Химия. Неорганическая химия. 8 класс. — 15-е изд. — М.: Просвещение, 2011. — С. 101. — ISBN 978-5-09-025532-5.
- ↑ Лидин Р. А., Молочко В. А., Андреева Л. Л., Цветков А. А. Основы номенклатуры неорганических веществ / Под ред. Б. Д. Степина. — М.: Химия, 1983. — С. 39—45.
- ↑ Степин Б. Д. , Цветков А. А. Неорганическая химия / Под ред. Б. Д. Степина. — М.: Высшая школа, 1994. — С. 18—19. — ISBN 5-06-001740-0.
- ↑ Кан Р., Дермер О. Введение в химическую номенклатуру = Introduction to Chemical Nomenclature / Пер. с англ. Н. Н. Щербиновской, под ред. В. М. Потапова и Р. А. Лидина. — М.: Химия, 1983. — С. 131—134.
- ↑ Терней А. Современная органическая химия. Т. 2. — М.: Мир, 1981. — С. 103.
- ↑ Senning A. Elsevier's Dictionary of Chemoetymology. The whies and whences of chemical nomenclature and terminology. — Elsevier, 2007. — 434 p. — ISBN 978-0-444-52239-9.
- ↑ Золотов и др., 1999, с. 121—122.
- ↑ 1 2 Рабинович В. А., Хавин З. Я. Краткий химический справочник. — Изд. 2-е, испр. и доп. — Ленинград: Химия, 1978. — С. 235.
- ↑ Химическая энциклопедия, 1992, т. 2, с. 396.
- ↑ 1 2 Дроздов А. А., Зломанов В. П. Химия элементов главных групп периодической системы Д.И.Менделеева: галогены. 9. Свойства оксокислот хлора (1998). Дата обращения: 17 апреля 2013. Архивировано 16 февраля 2013 года.
- ↑ Шрайвер, Эткинс, 2004, с. 244.
- ↑ Химическая энциклопедия, 1992, т. 5, с. 432.
- ↑ Levelling effect // IUPAC Glossary of terms used in physical organic chemistry. Pure & Appl. Chem., Vol. 51, p. 1768. Дата обращения: 24 апреля 2013. Архивировано из оригинала 4 октября 2013 года.
- ↑ Реутов и др., 2010, с. 230—232.
- ↑ Неорганическая химия / Под ред. Ю. Д. Третьякова. — М.: Академия, 2004. — Т. 1. — С. 89—94. — ISBN 5-7695-1446-9.
- ↑ Реутов и др., 2010, с. 231.
- ↑ 1 2 Золотов и др., 1999, с. 123—125.
- ↑ Танганов Б. Б. Химические методы анализа. — Улан-Удэ: Издательство ВСГТУ, 2005. — С. 8—14. — ISBN 5-89230-037-4. Архивировано 22 февраля 2016 года.
- ↑ 1 2 Реутов и др., 2010, с. 233.
- ↑ Kirill M Dyumaev, B A Korolev. The Influence of Solvation on the Acid-Base Properties of Organic Compounds in Various Media // Russian Chemical Reviews. — 1980-11-30. — Т. 49, вып. 11. — С. 1021–1032. — ISSN 1468-4837 0036-021X, 1468-4837. — doi:10.1070/rc1980v049n11abeh002521.
- ↑ 1 2 3 4 5 6 7 8 9 Марч Дж. Органическая химия. Реакции, механизмы и структура. Т. 1 / Пер. с англ. З. Е. Самойловой, под ред. И. П. Белецкой. — М.: Мир, 1987. — С. 340—346.
- ↑ 1 2 3 Штейнгарц В. Д. Суперкислоты // Соросовский образовательный журнал. — 1999. — № 3. — С. 82—87. Архивировано 14 июля 2014 года.
- ↑ IUPAC Gold Book — superacid. Дата обращения: 23 апреля 2013. Архивировано 28 апреля 2013 года.
- ↑ Мануйлов А. В., Родионов В. И. — Кислоты. Классификация кислот. Химические свойства. Архивная копия от 21 февраля 2010 на Wayback Machine // Основы химии. Интернет-учебник.
- ↑ Павел Оржековский, Людмила Мещерякова, Марина Шалашова. Химия. 9 класс. Учебник для общеобразовательных учреждений. — Дрофа. — Москва: Астрель, 2016. — С. 124.
Литература
- Реутов О. А., Курц А. Л., Бутин К. П. Органическая химия. — 3-е изд. — М.: Бином. Лаборатория знаний, 2010. — Т. 1. — 567 с. — ISBN 978-5-94774-613-6.
- Шрайвер Д., Эткинс П. Неорганическая химия = Inorganic Chemistry / Пер. с англ. М. Г. Розовой, С. Я. Истомина, М. Е. Тамм, под ред. В. П. Зломанова. — М.: Мир, 2004. — Т. 1. — 679 с. — ISBN 5-03-003628-8.
Ссылки
- Все о кислотах Архивная копия от 21 февраля 2010 на Wayback Machine
- Органические и неорганические кислоты Архивная копия от 5 сентября 2009 на Wayback Machine
- Кислоты: бескислородные и кислородосодержащие, соли бескислородных и кислородсодержащих кислот, типы химических реакций Архивная копия от 16 сентября 2013 на Wayback Machine