Контрольная точка веретена

Контрольная точка веретена, также известная как переход от метафазы к анафазе, контрольная точка сборки веретена (SAC), контрольная точка метафазы или митотическая контрольная точка, представляет собой контрольную точку клеточного цикла во время митоза или мейоза, которая предотвращает разделение дуплицированных хромосом (анафазу) до тех пор, пока каждая хромосома не будет должным образом прикреплена к веретену. Для достижения правильной сегрегации две кинетохоры на сестринских хроматидах должны быть прикреплены к противоположным полюсам веретена (биполярная ориентация)[1]. Только такой способ прикрепления гарантирует, что каждая дочерняя клетка получит одну копию хромосомы. Определяющей биохимической особенностью этой контрольной точки является стимуляция комплекса, способствующего анафазе, комплексами M-фазы циклин-CDK, что, в свою очередь, вызывает протеолитическую деструкцию циклинов и белков, удерживающих вместе сестринские хроматиды[2].
Обзор и значение
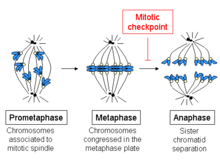
Начало метафазы характеризуется соединением микротрубочек с кинетохорами хромосом, а также выстраиванием хромосом в середине клетки. Каждая хроматида имеет свою собственную кинетохору, и все микротрубочки, связанные с кинетохорами сестринских хроматид, расходятся от противоположных полюсов клетки. Эти микротрубочки притягивают хромосомы к противоположным концам клеток, в то время как сцепление между сестринскими хроматидами противодействует этой силе.
При переходе от метафазы к анафазе эта связь между сестринскими хроматидами разрушается, и разделенные хроматиды вытягиваются к противоположным сторонам клетки с помощью микротрубочек веретена. Хроматиды далее разделяются физическим движением самих полюсов веретена. Преждевременная диссоциация хроматид может привести к неправильной сегрегации хромосом и анеуплоидии в дочерних клетках. Таким образом, задача контрольной точки веретена состоит в том, чтобы предотвратить этот переход в анафазу до тех пор, пока хромосомы не будут должным образом прикреплены, прежде чем сестринские хроматиды разделятся.
Чтобы сохранить идентичность клетки и её правильное функционирование, необходимо поддерживать соответствующее количество хромосом после каждого клеточного деления. Ошибка в создании дочерних клеток с меньшим или большим числом хромосом, чем ожидалось (ситуация, называемая анеуплоидией), может в лучшем случае привести к гибели клетки или, наоборот, может привести к катастрофическим фенотипическим результатам[3][4]. Примеры включают:
- В раковых клетках анеуплоидия является частым явлением, указывающим на то, что эти клетки представляют дефект в механизме, участвующем в сегрегации хромосом, а также в механизме, обеспечивающем правильное выполнение сегрегации.
- У человека синдром Дауна появляется у детей, несущих в своих клетках одну лишнюю копию хромосомы 21, в результате дефекта сегрегации хромосом во время мейоза у одного из предшественников. Этот дефект приведет к образованию гаметы (сперматозоида или ооцита) с дополнительной хромосомой 21. После оплодотворения эта гамета даст эмбрион с тремя копиями хромосомы 21.
Обнаружение контрольной точки сборки веретена (SAC)

Циркле (в 1970 г.) был одним из первых исследователей, обнаруживших, что, когда хотя бы одна хромосома задерживается в процессе прохождения пути до метафазной пластинки, начало анафазы откладывается на несколько минут после её прибытия[5]. Это наблюдение, вместе с аналогичными, предполагает, что существует механизм контроля при переходе от метафазы к анафазе. При использовании таких препаратов, как нокодазол и колхицин, происходит разборка митотического веретена и блокировка клеточного цикла при переходе от метафазы к анафазе. При использовании этих препаратов (см. обзор Ридера и Палаццо в 1992 г.[6]) предполагаемый механизм контроля получил название Spindle Assembly Checkpoint (SAC, контрольная точка веретена) . С тех пор этот регуляторный механизм интенсивно изучается[7].
С помощью различных видов генетических исследований установлено, что активировать SAC способны различного рода дефекты: деполимеризация веретена[8][9], наличие дицентрических хромосом (с двумя центромерами)[10], расхождение центромер в аберрантным путем[11], дефекты в телах полюсов веретена у S. cerevisiae[12], дефекты кинетохорных белков[13], мутации центромерной ДНК[14] или дефекты молекулярных моторов, активных во время митоза[8]. Краткое изложение этих наблюдений можно найти в статье Хардвика и его сотрудников в 1999 г.[15].
Используя собственные наблюдения, Циркле[5] впервые предположил, что «какое-то (…) вещество, необходимое клетке для перехода в анафазу, появляется через несколько минут после С (момент прибытия последней хромосомы на метафазную пластинку), или после резкого изменения цитоплазматического состояния, сразу в С или сразу после С», что позволяет предположить, что эта функция локализована на кинетохорах, не прикрепленных к митотическому веретену. Макинтош расширил это предположение, предполагая, что один фермент, чувствительный к натяжению, расположенный в центромерах, продуцирует ингибитор начала анафазы, когда две сестринские кинетохоры не находятся под биполярным натяжением[16]. Действительно, имеющиеся данные свидетельствуют о том, что сигнал «ожидание входа в анафазу» производится в основном на неприкрепленных кинетохорах или рядом с ними[17]. Однако первичным событием, связанным с присоединением кинетохоры к веретену, которое способно инактивировать ингибирующий сигнал и снять блокировку метафазы, может быть либо приобретение кинетохорой микротрубочек (как было предложено Ридером и сотрудниками в 1995 г.[17].), или натяжение, стабилизирующее заякоривание микротрубочек на кинетохорах (как предполагают эксперименты, проведенные в лаборатории Никласа[18]). Последующие исследования на клетках, содержащих два независимых митотических веретена в единственной цитоплазме, показали, что ингибитор перехода от метафазы к анафазе генерируется неприкрепленными кинетохорами и не диффундирует свободно в цитоплазме[19]. Тем не менее, в том же исследовании было показано, что, как только переход от метафазы к анафазе инициируется в одной части клетки, эта информация распространяется по всей цитоплазме и может преодолеть сигнал «подождите, чтобы войти в анафазу», связанный с переходом в анафазу. второе веретено, содержащее неприкрепленные кинетохоры.
Справочная информация о дупликации сестринских хроматид, сплоченности и сегрегации
Деление клеток: дублирование материала и распространение в дочерние клетки
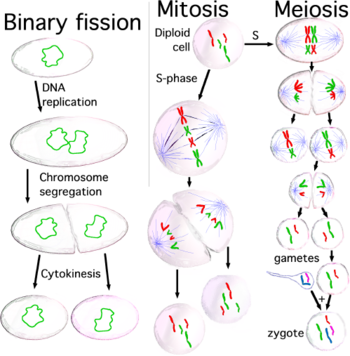
Когда клетки готовы к делению, поскольку их размер достаточно велик или они получают соответствующий стимул[20], они активируют механизм для вступления в клеточный цикл и дублируют большинство органелл во время фазы S (синтеза), включая их центросомы. Поэтому, когда процесс клеточного деления завершится, каждая дочерняя клетка получит полный набор органелл. В то же время во время S-фазы все клетки должны очень точно дублировать свою ДНК — процесс, называемый репликацией ДНК. Как только репликация ДНК завершена, у эукариот молекула ДНК уплотняется и конденсируется, образуя митотические хромосомы, каждая из которых состоит из двух сестринских хроматид, которые удерживаются вместе за счет установления сцепления между ними; каждая хроматида представляет собой законченную молекулу ДНК, прикрепленную через микротрубочки к одной из двух центросом делящейся клетки, расположенных на противоположных полюсах клетки. Структура, образованная центросомами и микротрубочками, называется митотическим веретеном из-за её характерной формы, удерживающей хромосомы между двумя центросомами. Обе сестринские хроматиды остаются вместе до анафазы; в этот момент они отделяются друг от друга и направляются к центросоме, к которой прикреплены. Таким образом, когда две дочерние клетки разделятся в конце процесса деления, каждая из них получит полный набор хроматид. Механизм, отвечающий за правильное распределение сестринских хроматид при клеточном делении, называется сегрегацией хромосом.
Чтобы обеспечить правильное расхождение хромосом, клетки разработали точный и сложный механизм. Во-первых, клетки должны координировать удвоение центросомы с репликацией ДНК, и сбой в этой координации будет генерировать монополярные или мультиполярные митотические веретена, которые обычно вызывают аномальное расхождение хромосом[21], потому что в этом случае распределение хромосом не будет происходить. сбалансированным образом.
Митоз: прикрепление хромосом к веретену деления и расхождение хромосом
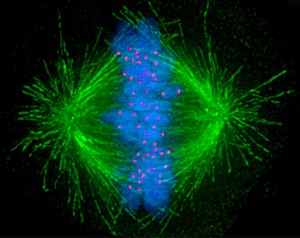
Во время S-фазы центросома начинает удваиваться. Как раз в начале митоза обе центриоли достигают максимальной длины, рекрутируют дополнительный материал и их способность образовывать ядра микротрубочек увеличивается. По мере прогрессирования митоза обе центросомы расходятся, образуя митотическое веретено[22]. Таким образом, митотическое веретено имеет два полюса, от которых исходят микротрубочки. Микротрубочки (МТ) представляют собой длинные белковые филаменты с асимметричными окончаниями: один конец, обозначенный как «минус» (-), относительно стабилен и близок к центросоме, а конец, обозначенный как «плюс» (+), с чередующимися фазами роста и ретракция, исследуя центр клетки в поисках хромосом. Каждая хроматида имеет особую область, называемую центромерой, поверх которой собрана белковая структура, называемая кинетохорой, которая способна стабилизировать плюс-конец микротрубочки. Поэтому, если случайно микротрубочка, исследующая центр клетки, встретится с кинетохорой, может случиться так, что кинетохора захватит её, так что хромосома прикрепится к веретену через кинетохору одной из своих сестринских хроматид. Хромосома играет активную роль в прикреплении кинетохор к веретену. С хроматином связан фактор обмена гуаниновых нуклеотидов (GEF), который стимулирует цитозольный Ran вблизи хромосомы для связывания GTP вместо GDP. Активированная GTP-связанная форма Ran высвобождает белки, стабилизирующие микротрубочки, такие как TPX2, из белковых комплексов в цитозоле, что вызывает зарождение и полимеризацию микротрубочек вокруг хромосом[2]. Эти микротрубочки, происходящие из кинетохор, наряду с кинезиновыми моторными белками во внешних кинетохорах, облегчают взаимодействие с латеральной поверхностью микротрубочек, происходящих из полюсов веретена. Однако эти боковые крепления нестабильны и должны быть преобразованы в крепление на конце. Преобразование латерального прикрепления в прикрепление концами позволяет преобразовать рост и сжатие плюс-концов микротрубочек в силы, толкающие и тянущие хромосомы для достижения правильной биориентации. Так как бывает, что сестринские хроматиды соединены вместе и обе кинетохоры расположены спина к спине на обеих хроматидах, когда одна кинетохора присоединяется к одной центросоме, сестринская кинетохора становится открытой для центросомы, расположенной на противоположном полюсе; по этой причине в большинстве случаев вторая кинетохора становится связанной с центросомой на противоположном полюсе через её микротрубочки[23], так что хромосомы становятся «двунаправленными», что является фундаментальной конфигурацией (также называемой амфителической), гарантирующей, что хромосома сегрегация будет происходить правильно, когда клетка будет делиться[24][25]. Иногда одна из двух сестринских кинетохор может одновременно прикрепляться к MTs, генерируемым обоими полюсами, конфигурация, названная меротелической, которая не обнаруживается контрольной точкой веретена, но может генерировать отстающие хромосомы во время анафазы и, следовательно, анеуплоидию. Меротелическая ориентация (характеризующаяся отсутствием напряжения между сестринскими кинетохорами) часто встречается в начале митоза, но белок Aurora B (киназа, сохранившаяся от дрожжей до позвоночных) обнаруживает и устраняет этот тип закрепления[26]. (Примечание: Aurora B часто гиперэкспрессируется в различных типах опухолей и в настоящее время является мишенью для разработки противоопухолевых препаратов[27].)
Слипание сестринских хроматид во время митоза
Когезин: белки SMC
Как было отмечено ранее, сестринские хроматиды остаются связанными с S-фазы (когда ДНК реплицируется с образованием двух идентичных копий, двух хроматид) до анафазы. В этот момент две сестринские хроматиды расходятся и расходятся к противоположным полюсам делящейся клетки. Генетические и биохимические исследования дрожжей и экстрактов яиц у Xenopus laevis идентифицировали полипротеиновый комплекс как существенный игрок в слипчивости сестринских хроматид (см. обзор Hirano в 2000[28]). Этот комплекс известен как когезиновый комплекс и у Saccharomyces cerevisiae состоит по меньшей мере из четырёх субъединиц: Smc1p, Smc3p, Scc1p (или Mcd1p) и Scc3p. И Smc1p, и Smc3p принадлежат к семейству белков для структурного поддержания хромосом (SMC), которые составляют группу высококонсервативных хромосомных АТФаз и образуют гетеродимер (Smc1p/Smc3p). Scc1p является гомологом Rad21 у S.cerevisiae, впервые идентифицированным как белок, участвующий в репарации ДНК у S.pombe . Эти четыре белка необходимы для дрожжей, и мутация в любом из них приведет к преждевременному расхождению сестринских хроматид. У дрожжей когезин связывается с предпочтительными участками вдоль плеч хромосом и очень обилен вблизи центромер, как это было показано в исследовании с использованием иммунопреципитации хроматина[29].
Роль гетерохроматина
Классические цитологические наблюдения подтвердили, что сестринские хроматиды более сильно прикреплены к гетерохроматическим областям[30], и это указывает на то, что особая структура или состав гетерохроматина может благоприятствовать рекрутированию когезин[31]. Фактически было показано, что Swi6 (гомолог HP-1 у S. pombe) связывается с метилированным Lys 9 гистона H3 и способствует связыванию когезина с центромерными повторами у S. pombe[32][33]. Более поздние исследования показывают, что механизм RNAi регулирует установление гетерохроматина, который, в свою очередь, рекрутирует когезин в эту область, как в S. pombe[34], так и в клетках позвоночных[35]. Однако должны быть другие механизмы, кроме гетерохроматина, для обеспечения усиленной когезии на центромерах, потому что у S. cerevisiae отсутствует гетерохроматин рядом с центромерами, но присутствие функциональной центромеры индуцирует увеличение когезиновой ассоциации в смежной области, охватывающей 20-50 т.п.н.[36]
В этом направлении Orc2 (один белок, включенный в комплекс распознавания источника, ORC, участвующий в инициации репликации ДНК во время S фазы) также расположен на кинетохорах во время митоза в клетках человека[37]; в согласии с этой локализацией, некоторые наблюдения показывают, что Orc2 у дрожжей участвует в слипании сестринских хроматид, и его удаление индуцирует активацию SAC[38]. Также было замечено, что др. компоненты комплекса ORC (такие как orc5 у S. pombe) участвуют в когезии. Однако молекулярный путь с участием белков ORC, по-видимому, дополняет путь когезинов и в основном неизвестен.
Функция сплоченности и её растворение
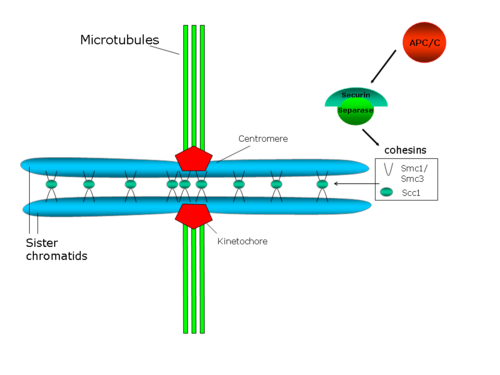
Центромерное сцепление сопротивляется силам, прилагаемым микротрубочками веретена к полюсам, которые создают напряжение между сестринскими кинетохорами. В свою очередь, это напряжение стабилизирует соединение микротрубочки-кинетохоры посредством механизма с участием белка Aurora B (обзор по этому вопросу : Хауф и Ватанабэ, 2004[39]).
Действительно, снижение клеточных уровней когезина приводит к преждевременному расхождению сестринских хроматид, а также к дефектам хромосомной конгрессии на метафазной пластинке и делокализации белков в хромосомном комплексе-пассажире, который содержит белок Aurora B[40][41]. Предлагаемая структура комплекса когезина предполагает, что этот комплекс непосредственно соединяет обе сестринские хроматиды[42]. В этой предполагаемой структуре SMC-компоненты когезина играют структурную роль, так что гетеродимер SMC может функционировать как ДНК-связывающий белок, конформация которого регулируется АТФ[43]. Однако Scc1p и Scc3p будут играть регулирующую роль[28].
У S. cerevisiae Pds1p (также известный как секурин) регулирует сцепление сестринских хроматид, поскольку он связывает и ингибирует протеазу Esp1p (сепарин или сепараза). Когда начинается анафаза, активирующий анафазу комплекс (APC/C или циклосома) расщепляет секурин. APC/C представляет собой кольцевую убиквитинлигазу Е3, которая рекрутирует убиквитин-конъюгирующий фермент Е2, нагруженный убиквитином. Секурин распознается только в том случае, если Cdc20, субъединица активатора, связана с ядром APC/C. Когда секурин, Cdc20 и E2 все связаны с APC/C, E2 убиквитинирует секурин и избирательно разрушает его. Деградация секурина высвобождает протеазу Esp1p/separase, которая разрушает когезиновые кольца, связывающие две сестринские хроматиды, тем самым способствуя разделению сестринских хроматид[44]. Также было показано, что киназа Polo/Cdc5 фосфорилирует остатки серина рядом с сайтом разрезания для Scc1, и это фосфорилирование должно способствовать активности разрезания[45].
Хотя этот механизм сохраняется в ходе эволюции[46][47], у позвоночных большинство молекул когезина высвобождается в профазе, независимо от присутствия APC/C, в процессе, зависящем от Polo-подобного 1 (PLK1) и Aurora B[48]. Однако было показано, что небольшое количество Scc1 остается связанным с центромерами в клетках человека до метафазы, и такое же количество вырезается в анафазе, когда он исчезает из центромер[49]. С другой стороны, некоторые эксперименты показывают, что сцепление сестринских хроматид в плечах постепенно утрачивается после разделения сестринских центромер, и сестринские хроматиды перемещаются к противоположным полюсам клетки[50][51].
По некоторым наблюдениям, часть когезинов хромосомных плеч и центромерных когезинов защищены белком шугошином (Shugoshin, Sgo1), избегая их высвобождения в профазе[52][53]. Чтобы иметь возможность функционировать в качестве защитника центромерной когезии, Sgo1 должен быть инактивирован в начале анафазы, так же как и Pds1p. Фактически, как Pds1p, так и Sgo1 являются субстратами APC/C у позвоночных[54].
Обзор контрольной точки веретена
Контрольная точка сборки веретена (SAC) представляет собой активный сигнал, производимый неправильно прикрепленными кинетохорами, который сохраняется у всех эукариот. SAC останавливает клеточный цикл, негативно регулируя CDC20, тем самым предотвращая активацию активности полиубиквитинирования комплекса, стимулирующего анафазу (APC). Белки, ответственные за сигнал SAC, составляют комплекс митотических контрольных точек (MCC), который включает белки SAC, MAD2/MAD3 (дефицит митотической остановки), BUB3 (почкование, не ингибируемое бензимидазолом) и CDC20[55]. Другие белки, участвующие в SAC, включают MAD1, BUB1, MPS1 и Aurora B. Для высших эукариот дополнительными регуляторами SAC являются составляющие комплекса ROD-ZW10, <a href="https://en.wikipedia.org/wiki/P31comet" rel="mw:ExtLink" title="P31comet" class="new cx-link" data-linkid="208">p31<sup>comet</sup></a>, MAPK, CDK1-циклин-B, NEK2 и PLK1[56].

Активация контрольной точки
SAC отслеживает взаимодействие между неправильно соединенными кинетохорами и микротрубочками веретена и поддерживается до тех пор, пока кинетохоры не будут должным образом прикреплены к веретену. Во время прометафазы CDC20 и белки SAC концентрируются на кинетохорах до прикрепления к сборке веретена. Эти белки поддерживают активацию SAC до тех пор, пока они не будут удалены и не будет установлено правильное прикрепление кинетохор к микротрубочкам. Даже одна неприкрепленная кинетохора может поддерживать контрольную точку веретена[55]. После прикрепления плюс-концов микротрубочек и образования кинетохорных микротрубочек MAD1 и MAD2 истощаются из сборки кинетохор. Другим регулятором активации контрольной точки является напряжение кинетохор. Когда сестринские кинетохоры должным образом прикреплены к противоположным полюсам веретена, силы в митотическом веретене создают напряжение в кинетохорах. Биориентированные сестринские кинетохоры стабилизируют сборку кинетохора-микротрубочки, тогда как слабое натяжение оказывает дестабилизирующее действие. В ответ на неправильное прикрепление кинетохор, такое как синтетическое прикрепление, когда обе кинетохоры прикрепляются к одному полюсу веретена, возникающее слабое натяжение дестабилизирует неправильное прикрепление и позволяет кинетохоре правильно прикрепиться к телу веретена. Во время этого процесса кинетохоры, прикрепленные к митотическому веретену, но не находящиеся под напряжением, запускают контрольную точку веретена. Киназа Aurora-B/Ipl1 хромосомного комплекса-пассажира функционирует как датчик натяжения в неправильных прикреплениях кинетохор. Он обнаруживает и дестабилизирует неправильные прикрепления посредством контроля кинезина KINI MCAK, разъединяющего микротрубочки, комплекса DASH и комплекса Ndc80/Hec1[57] на границе микротрубочки-кинетохора[56]. Киназа Aurora-B/Ipl1 также имеет решающее значение для коррекции меротелических прикреплений, когда один кинетохор одновременно прикрепляется к обоим полюсам веретена. Меротелиальные прикрепления создают достаточное напряжение и не обнаруживаются SAC, а без коррекции могут привести к неправильной сегрегации хромосом из-за низкой скорости миграции хроматид. В то время как прикрепление микротрубочек независимо необходимо для активации SAC, неясно, является ли натяжение независимым регулятором SAC, хотя ясно, что при натяжении возникают различные регуляторные поведения.
После активации контрольная точка веретена блокирует вход в анафазу, ингибируя комплекс, способствующий анафазе, посредством регуляции активности комплекса митотической контрольной точки. Механизм ингибирования APC комплексом митотических контрольных точек плохо изучен, хотя предполагается, что MCC связывается с APC как псевдосубстрат, используя мотив KEN-box в BUBR1. В то же время, когда активируется комплекс митотических контрольных точек, белок центромер CENP-E активирует BUBR1, который также блокирует анафазу[56].
Формирование комплекса митотических контрольных точек
Комплекс митотической контрольной точки состоит из BUB3 вместе с MAD2 и MAD3, связанными с Cdc20. MAD2 и MAD3 имеют разные сайты связывания на CDC20 и действуют синергически, ингибируя APC/C. Комплекс MAD3 состоит из BUB3, который связывается с Mad3 и BUB1B посредством короткого линейного мотива, известного как мотив GLEBS. Точный порядок прикрепления, который должен иметь место для формирования MCC, остается неизвестным. Возможно, что Mad2-Cdc20 образуют комплекс в то же самое время, когда BUBR1-BUB3-Cdc20 образуют другой комплекс, и эти два субкомплекса, следовательно, объединяются в комплекс митотической контрольной точки[55]. В клетках человека для связывания BUBR1 с CDC20 требуется предварительное связывание MAD2 с CDC20, поэтому возможно, что субкомплекс MAD2-CDC20 действует как инициатор образования MCC. Истощение BUBR1 приводит только к умеренному снижению уровней Mad2-Cdc20, в то время как Mad2 необходим для связывания BubR1-Bub3 с Cdc20. Тем не менее, BUBR1 по-прежнему требуется для активации контрольной точки[56].
Механизм образования MCC неясен, и существуют конкурирующие теории как для кинетохор-зависимого, так и для кинетохор-независимого образования. В поддержку теории, не зависящей от кинетохор, MCC обнаруживается в клетках S. cerevisiae, в которых белки сборки ядра кинетокора были мутированы, и в клетках, в которых SAC был дезактивирован, что предполагает, что MCC может собираться во время митоза без локализации кинетохор. В одной модели неприкрепленные прометафазные кинетохоры могут «сенсибилизировать» APC к ингибированию MCC путем привлечения APC к кинетохорам через функционирующий SAC. Кроме того, истощение различных белков SAC показало, что истощение MAD2 и BUBR1 влияет на время митоза независимо от кинетохоров, в то время как истощение других белков SAC приводит к дисфункциональным SAC без изменения продолжительности митоза. Таким образом, возможно, что SAC функционирует через двухэтапный таймер, где MAD2 и BUBR1 контролируют продолжительность митоза на первой стадии, которая может увеличиваться на второй стадии, если есть неприкрепленные кинетохоры, а также другие белки SAC[56]. Однако есть ряд свидетельств, которые не в пользу независимой от кинетохор сборки. MCC ещё предстоит обнаружить во время интерфазы, в то время как MCC не образуется из своих компонентов в экстрактах мейоза II X. laevis без добавления сперматозоидов ядер и нокодазола для предотвращения сборки веретена.
Ведущей моделью формирования MCC является «модель шаблона MAD2», которая зависит от динамики кинетохор MAD2 для создания MCC. MAD1 локализуется на неприкрепленных кинетохорах, при этом сильно связываясь с MAD2. Локализация MAD2 и BubR1 на кинетохоре также может зависеть от киназы Aurora B[58]. Клетки, лишенные Aurora B, не могут останавливаться в метафазе, даже если в хромосомах отсутствует прикрепление микротрубочек[59]. Неприкрепленные кинетохоры сначала связываются с комплексом MAD1-C-MAD2-p31comet и высвобождают p31comet посредством неизвестных механизмов. Образовавшийся комплекс MAD-C-MAD2 рекрутирует открытый конформер Mad2 (O-Mad2) на кинетохоры. Этот O-Mad2 меняет свою конформацию на закрытую Mad2 (C-Mad2) и связывает Mad1. Этот комплекс Mad1/C-Mad2 отвечает за рекрутирование большего количества O-Mad2 в кинетохоры, которые изменяют свою конформацию на C-Mad2 и связывают Cdc20 в реакции автоамплификации. Поскольку MAD1 и CDC20 содержат сходный MAD2-связывающий мотив, пустая конформация O-MAD2 изменяется на C-MAD2 при связывании с CDC20. Эта петля положительной обратной связи отрицательно регулируется p31comet, которая конкурентно связывается с C-MAD2, связанным либо с MAD1, либо с CDC20, и снижает дальнейшее связывание O-MAD2 с C-MAD2. Также могут существовать дополнительные механизмы контроля, учитывая, что p31comet отсутствует у низших эукариот. Таким образом, номенклатура «шаблонной модели» происходит от процесса, в котором MAD1-C-MAD2 действует как шаблон для формирования копий C-MAD2-CDC20. Эта секвестрация Cdc20 необходима для поддержания контрольной точки веретена[55].
Деактивация контрольных точек
Существует несколько механизмов дезактивации SAC после правильной биориентации сестринских хроматид. При прикреплении микротрубочек к кинетохоре механизм отщепления через моторный комплекс динеин-динеин транспортирует белки контрольных точек веретена от кинетохор[56]. Удаленные белки, которые включают MAD1, MAD2, MPS1 и CENP-F, затем перераспределяются на полюса веретена . Процесс удаления сильно зависит от неповрежденной структуры микротрубочек, а также от подвижности динеина вдоль микротрубочек. Помимо функции регулятора петли положительной обратной связи C-MAD2, p31comet также может действовать как деактиватор SAC. Неприкрепленные кинетохоры временно инактивируют p31comet, но прикрепление повторно активирует белок и ингибирует активацию MAD2, возможно, путем ингибирующего фосфорилирования. Другой возможный механизм инактивации SAC возникает в результате энергозависимой диссоциации комплекса MAD2-CDC20 посредством недеградационного убиквитинирования CDC20. И наоборот, деубиквитилирующий фермент протектин необходим для поддержания SAC. Таким образом, неприкрепленные кинетохоры поддерживают контрольную точку, непрерывно воссоздавая субкомплекс MAD2-CDC20 из его компонентов. SAC также может быть дезактивирован протеолизом, индуцированным активацией APC. Поскольку SAC не реактивируется при потере сцепления сестринских хроматид во время анафазы, протеолиз циклина B и инактивация CDK1-циклин-B киназы также ингибируют активность SAC. Деградация MPS1 во время анафазы предотвращает реактивацию SAC после удаления сцепления сестринских хроматид. После деактивации контрольной точки и во время нормальной анафазы клеточного цикла комплекс, стимулирующий анафазу, активируется за счет снижения активности MCC. Когда это происходит, ферментный комплекс полиубиквитинирует ингибитор анафазы секурин. Убиквитинирование и разрушение секурина в конце метафазы высвобождает активную протеазу, называемую сепаразой. Сепараза расщепляет молекулы когезии, которые удерживают сестринские хроматиды вместе, чтобы активировать анафазу[2].
Новая модель дезактивации SAC у S. cerevisiae: механический переключатель
Был предложен новый механизм для объяснения того, как прикрепление микротрубочек концами к кинетохоре способно нарушать специфические этапы передачи сигналов SAC. В неприкрепленной кинетохоре первым этапом образования MCC является фосфорилирование Spc105 киназой Mps1. Затем фосфорилированный Spc105 способен рекрутировать нижестоящие сигнальные белки Bub1 и 3; Mad 1,2 и 3; и Cdc20. Ассоциация с Mad1 на неприкрепленных кинетохорах заставляет Mad2 претерпевать конформационные изменения, которые превращают его из открытой формы (O-Mad2) в закрытую форму (C-Mad2.) C-Mad2, связанный с Mad1, затем димеризуется со вторым O-Mad2 и катализирует его замыкание вокруг Cdc20. Этот комплекс C-Mad2 и Cdc20, MCC, оставляет Mad1 и C-Mad2 на кинетохоре, чтобы сформировать другой MCC. Каждый MCC изолирует две молекулы Cdc20, чтобы предотвратить их взаимодействие с APC/C, тем самым поддерживая SAC[2]. Фосфорилирование Spc105 с помощью Mps1 необходимо и достаточно для инициации пути передачи сигналов SAC, но эта стадия может происходить только в отсутствие прикрепления микротрубочек к кинетохоре. Показано, что эндогенный Mps1 ассоциирован с доменом кальпонин-гомологии (calponin-homology, CH) Ndc80, который расположен во внешней кинетохорной области, удаленной от хромосомы. Хотя Mps1 закреплен во внешней кинетохоре, он все же способен локализоваться во внутренней кинетохоре и фосфорилировать Spc105 из-за гибких шарнирных областей на Ndc80. Однако модель механического переключателя предполагает, что присоединение микротрубочки к концу кинетохоры деактивирует SAC посредством двух механизмов. Присутствие прикрепленной микротрубочки увеличивает расстояние между СН-доменом Ndc80 и Spc105. Кроме того, Dam1/DASH, большой комплекс, состоящий из 160 белков, который образует кольцо вокруг прикрепленной микротрубочки, действует как барьер между двумя белками. Разделение предотвращает взаимодействие между Mps1 и Spc105 и, таким образом, ингибирует сигнальный путь SAC[60].
Эта модель неприменима к регуляции SAC у организмов более высокого порядка, включая животных. Основным аспектом механизма механического переключения является то, что у S. cerevisiae структура кинетохоры позволяет прикреплять только одну микротрубочку. Кинетохоры у животных, с другой стороны, представляют собой гораздо более сложные сети, содержащие сайты связывания для множества микротрубочек[61]. Прикрепление микротрубочек ко всем местам связывания кинетохор не обязательно для деактивации SAC и перехода в анафазу. Следовательно, в кинетохоре животных сосуществуют состояния с прикреплением к микротрубочкам и без прикрепления к микротрубочкам, в то время как SAC ингибируется. Эта модель не включает барьер, который препятствовал бы Mps1, связанному с прикрепленной кинетохорой, от фосфорилирования Spc105 в соседней неприкрепленной кинетохоре. Кроме того, дрожжевой комплекс Dam1/DASH отсутствует в клетках животных.
Дефекты контрольной точки веретена и рак
Когда контрольная точка веретена неправильно функционирует, это может привести к неправильной сегрегации хромосом, анеуплоидии и даже онкогенезу[56]. Трансформация происходит и ускоряется, когда нарушается поддержание целостности генома, особенно на общем уровне целых хромосом или их больших частей. Фактически, анеуплоидия является наиболее распространенной характеристикой солидных опухолей человека, и поэтому контрольная точка сборки веретена может рассматриваться как возможная мишень для противоопухолевой терапии[62]. Это очень недооцененный факт, поскольку мутации в определённых генах, известных как онкогены или супрессоры опухолей, в первую очередь считаются причиной генетической нестабильности и онкогенеза. Обычно различные контрольные точки в клеточном цикле обеспечивают целостность генома посредством высококонсервативных избыточных механизмов, которые важны для поддержания клеточного гомеостаза и предотвращения онкогенеза. Несколько белков контрольных точек сборки веретена действуют как положительные и отрицательные регуляторы, чтобы обеспечить правильное расхождение хромосом в каждом клеточном цикле, предотвращая нестабильность хромосом (CIN), также известную как нестабильность генома.

В настоящее время целостность генома оценивается на нескольких уровнях, где некоторые опухоли проявляют нестабильность, проявляющуюся в виде замен оснований, вставок и делеций, в то время как в большинстве случаев наблюдается увеличение или потеря целых хромосом[63].
Из-за того, что изменения в митотических регуляторных белках могут приводить к анеуплоидии, а это частое явление при раке[64], первоначально предполагалось, что эти гены могут мутировать в раковых тканях[65].
Мутированные гены при раке
При некоторых видах рака хорошо охарактеризованы гены, лежащие в основе дефектов, приводящих к трансформации. При гематологическом раке, таком как множественная миелома, цитогенетические аномалии очень распространены из-за врожденной природы разрывов ДНК, необходимых для перестройки гена иммуноглобулина. Однако дефекты в белках, таких как MAD2, которые функционируют преимущественно в SAC, также характерны для множественной миеломы[66]. Большинство твёрдых опухолей также преимущественно анеуплоидны. Для колоректального рака BUB1 и BUBR1, а также амплификация STK15 являются ключевыми регуляторами, которые вовлечены в геномную нестабильность, приводящую к раку[67]. При раке молочной железы генетическая форма, характеризующаяся геном BRCA-1, демонстрирует более высокий уровень геномной нестабильности, чем спорадические формы. Эксперименты показали, что у мышей с нулевым геном BRCA-1 снижена экспрессия ключевого белка контрольной точки веретена MAD2[68]. Для других видов рака требуется дополнительная работа по выявлению причин анеуплоидии.
Другие гены, традиционно не связанные с SAC при раке
Очевидно, что вариации физиологических уровней этих белков (таких как Mad2 или BubR1) связаны с анеуплоидией и онкогенезом, и это было продемонстрировано на животных моделях[69][70]. Однако недавние исследования показывают, что то, что, по-видимому, происходит, является более сложным сценарием: анеуплоидия может привести к высокой частоте онкогенеза только тогда, когда изменения в уровнях специфических компонентов митотических контрольных точек (либо снижение, либо избыточная экспрессия) в тканях также индуцируют другие дефекты, способные предрасполагают их к опухолям[71]. То есть дефекты, такие как увеличение повреждения ДНК, хромосомные перестройки и/или снижение частоты гибели клеток. Для некоторых компонентов митотических контрольных точек известно, что они участвуют в функциях вне митоза: ядерный импорт (Mad1), репрессия транскрипции (Bub3) и гибель клеток, ответ на повреждение ДНК, старение и мегакариопоэз для BubR1. Все это подтверждает вывод о том, что усиление туморогенеза связано не только с анеуплоидией, но и с другими дефектами[71].
Связанные с раком мутации, затрагивающие известные гены контрольных точек, такие как BUB1 или BUBR1, на самом деле редки. Однако несколько белков, вовлеченных в рак, пересекаются с сетями сборки веретена. Ключевые супрессоры опухолей, такие как p53, также играют роль в контрольной точке веретена. Отсутствие p53, наиболее часто мутирующего гена при раке человека, оказывает большое влияние на регуляторы контрольных точек клеточного цикла, и в прошлом было показано, что он действует на контрольные точки G1, но теперь, по-видимому, также важен для регуляции контрольных точек веретена[72]. Другим ключевым аспектом рака является ингибирование гибели клеток или апоптоза. Сурвивин, член семейства ингибиторов апоптоза (IAP), локализован в пулах микротрубочек митотического веретена вблизи центросом и на кинетохорах метафазных хромосом. Мало того, что сурвивин ингибирует апоптоз, чтобы способствовать онкогенезу, но он был вовлечен (через экспериментальных мышей с нокаутом) в качестве важного регулятора сегрегации хромосом и поздних стадий митоза, аналогично его роли в более примитивных организмах[73].
Др. аспекты контрольной точки сборки веретена, такие как прикрепление кинетохор, функция микротрубочек и слипчивость сестринских хроматид, скорее всего, также дефектны и вызывают анеуплоидию. Было замечено, что раковые клетки делятся в нескольких направлениях, уклоняясь от контрольной точки сборки веретена, что приводит к мультиполярным митозам[74]. Мультиполярный переход метафаза-анафаза происходит через неполный цикл сепаразы, что приводит к частым событиям нерасхождения, которые усиливают анеуплоидию в раковых клетках.
Лечение рака SAC

Достижения в этой области привели к внедрению некоторых методов лечения, нацеленных на дефекты сборки веретена. Старые методы лечения, такие как алкалоиды барвинка и таксаны, нацелены на микротрубочки, которые сопровождают образование митотического веретена, путем нарушения динамики микротрубочек, которые задействуют SAC, останавливая клетку и в конечном итоге приводя к её гибели[75]. Таксол и доцетаксел до сих пор используются для лечения рака молочной железы, рака яичников и других видов эпителиального рака. Однако эти методы лечения часто характеризуются высокой частотой побочных эффектов и лекарственной устойчивостью.
Другие цели в сети регулирующих органов, влияющих на SAC, также преследуются; большой интерес сместился в сторону белков киназы aurora[76]. Ген киназы Aurora A при амплификации действует как онкоген, подавляющий SAC, что приводит к аномальному инициированию анафазы и последующей анеуплоидии, а также к резистентности к TAXOL[77]. Интересно, что низкомолекулярный ингибитор Aurora A продемонстрировал противоопухолевый эффект в модели in vivo, что позволяет предположить, что он может стать хорошей целью для дальнейшей клинической разработки[78]. Ингибиторы Aurora B, которые также находятся в стадии клинической разработки, приводят к аномальному прикреплению кинетохор к микротрубочкам, а также отменяют контрольную точку митоза[76]. Сурвивин также является привлекательной молекулярной мишенью для клинической терапевтической разработки, поскольку он действует как основной узел во множестве путей, одним из которых является формирование веретена и контроль контрольных точек[79]. Даже дальнейшие подходы включали ингибирование митотических моторных белков, таких как KSP. Эти ингибиторы, которые недавно прошли клинические испытания, вызывают остановку митоза и, задействуя контрольную точку сборки веретена, индуцируют апоптоз[80][3].
Примечания
- ↑ "The life and miracles of kinetochores". The EMBO Journal. 28 (17): 2511—31. September 2009. doi:10.1038/emboj.2009.173. PMID 19629042.
- ↑ 1 2 3 4 David Owen Morgan. The cell cycle : principles of control. — London: New Science Press, 2007. — xxvii, 297 pages с. — ISBN 978-0-19-920610-0, 0-19-920610-4, 978-0-9539181-2-6, 0-9539181-2-2, 978-0-87893-508-6, 0-87893-508-8.
- ↑ 1 2 Cell Cycle
{{citation}}:|title=пропущен или пуст () - ↑ "Short- and long-term effects of chromosome mis-segregation and aneuploidy". Nature Reviews Molecular Cell Biology. 16 (8): 473—85. August 2015. doi:10.1038/nrm4025. PMID 26204159.
- ↑ 1 2 "Ultraviolet-microbeam irradiation of newt-cell cytoplasm: spindle destruction, false anaphase, and delay of true anaphase". Radiation Research. 41 (3): 516—37. March 1970. Bibcode:1970RadR...41..516Z. doi:10.2307/3572841. PMID 5438206.
- ↑ "Colcemid and the mitotic cycle". Journal of Cell Science. 102 ( Pt 3) (3): 387—92. July 1992. doi:10.1242/jcs.102.3.387. PMID 1506421.
- ↑ "Linking kinetochore-microtubule binding to the spindle checkpoint". Developmental Cell. 14 (4): 474—9. April 2008. doi:10.1016/j.devcel.2008.03.015. PMID 18410725.
- ↑ 1 2 "Feedback control of mitosis in budding yeast". Cell. 66 (3): 519—31. August 1991. doi:10.1016/0092-8674(81)90015-5. PMID 1651172.
- ↑ "S. cerevisiae genes required for cell cycle arrest in response to loss of microtubule function". Cell. 66 (3): 507—17. August 1991. doi:10.1016/0092-8674(81)90014-3. PMID 1651171.
- ↑ "A delay in the Saccharomyces cerevisiae cell cycle that is induced by a dicentric chromosome and dependent upon mitotic checkpoints". Molecular and Cellular Biology. 12 (9): 3857—64. September 1992. doi:10.1128/MCB.12.9.3857. PMID 1324407.
- ↑ "Aberrantly segregating centromeres activate the spindle assembly checkpoint in budding yeast". The Journal of Cell Biology. 133 (1): 75—84. April 1996. doi:10.1083/jcb.133.1.75. PMID 8601615.
- ↑ "Activation of the budding yeast spindle assembly checkpoint without mitotic spindle disruption". Science. 273 (5277): 953—6. August 1996. Bibcode:1996Sci...273..953H. doi:10.1126/science.273.5277.953. PMID 8688079.
- ↑ "Checkpoint genes required to delay cell division in response to nocodazole respond to impaired kinetochore function in the yeast Saccharomyces cerevisiae". Molecular and Cellular Biology. 15 (12): 6838—44. December 1995. doi:10.1128/MCB.15.12.6838. PMID 8524250.
- ↑ "Centromere DNA mutations induce a mitotic delay in Saccharomyces cerevisiae". Proceedings of the National Academy of Sciences of the United States of America. 89 (19): 8908—12. October 1992. Bibcode:1992PNAS...89.8908S. doi:10.1073/pnas.89.19.8908. PMID 1409584.
- ↑ "Lesions in many different spindle components activate the spindle checkpoint in the budding yeast Saccharomyces cerevisiae". Genetics. 152 (2): 509—18. June 1999. doi:10.1093/genetics/152.2.509. PMID 10353895.
- ↑ "Structural and mechanical control of mitotic progression". Cold Spring Harbor Symposia on Quantitative Biology. 56: 613—9. 1991. doi:10.1101/sqb.1991.056.01.070. PMID 1819511.
- ↑ 1 2 "The checkpoint delaying anaphase in response to chromosome monoorientation is mediated by an inhibitory signal produced by unattached kinetochores". The Journal of Cell Biology. 130 (4): 941—8. August 1995. doi:10.1083/jcb.130.4.941. PMID 7642709.
- ↑ "Tension-sensitive kinetochore phosphorylation and the chromosome distribution checkpoint in praying mantid spermatocytes". Journal of Cell Science. 110 ( Pt 5) (5): 537—45. March 1997. doi:10.1242/jcs.110.5.537. PMID 9092936.
- ↑ "Mitosis in vertebrate somatic cells with two spindles: implications for the metaphase/anaphase transition checkpoint and cleavage". Proceedings of the National Academy of Sciences of the United States of America. 94 (10): 5107—12. May 1997. Bibcode:1997PNAS...94.5107R. doi:10.1073/pnas.94.10.5107. PMID 9144198.
- ↑ "Size control in animal development". Cell. 96 (2): 235—44. January 1999. doi:10.1016/S0092-8674(00)80563-2. PMID 9988218.
- ↑ "Centrosome duplication in mammalian somatic cells requires E2F and Cdk2-cyclin A". Nature Cell Biology. 1 (2): 88—93. June 1999. doi:10.1038/10054. PMID 10559879.
- ↑ "Protein kinases in control of the centrosome cycle". FEBS Letters. 452 (1—2): 92—5. June 1999. doi:10.1016/S0014-5793(99)00534-7. PMID 10376685.
- ↑ "How cells get the right chromosomes". Science. 275 (5300): 632—7. January 1997. doi:10.1126/science.275.5300.632. PMID 9005842.
- ↑ "The centromere geometry essential for keeping mitosis error free is controlled by spindle forces". Nature. 450 (7170): 745—9. November 2007. Bibcode:2007Natur.450..745L. doi:10.1038/nature06344. PMID 18046416.
- ↑ "Tension between two kinetochores suffices for their bi-orientation on the mitotic spindle". Nature. 428 (6978): 93—7. March 2004. Bibcode:2004Natur.428...93D. doi:10.1038/nature02328. PMID 14961024.
- ↑ "Aurora kinase promotes turnover of kinetochore microtubules to reduce chromosome segregation errors". Current Biology. 16 (17): 1711—8. September 2006. doi:10.1016/j.cub.2006.07.022. PMID 16950108.
- ↑ "Aurora kinases as anticancer drug targets". Clinical Cancer Research. 14 (6): 1639—48. March 2008. doi:10.1158/1078-0432.CCR-07-2179. PMID 18347165.
- ↑ 1 2 "Chromosome cohesion, condensation, and separation". Annual Review of Biochemistry. 69: 115—44. 2000. doi:10.1146/annurev.biochem.69.1.115. PMID 10966455.
- ↑ "Establishment and maintenance of sister chromatid cohesion in fission yeast by a unique mechanism". The EMBO Journal. 20 (20): 5779—90. October 2001. doi:10.1093/emboj/20.20.5779. PMID 11598020.
- ↑ "The spindle is required for the process of sister chromatid separation in Drosophila neuroblasts". Experimental Cell Research. 192 (1): 10—5. January 1991. doi:10.1016/0014-4827(91)90150-S. PMID 1898588.
- ↑ "Shaping the metaphase chromosome: coordination of cohesion and condensation". BioEssays. 23 (10): 924—35. October 2001. doi:10.1002/bies.1133. PMID 11598959.
- ↑ "Requirement of heterochromatin for cohesion at centromeres". Science. 294 (5551): 2539—42. December 2001. Bibcode:2001Sci...294.2539B. doi:10.1126/science.1064027. PMID 11598266.
- ↑ "Recruitment of cohesin to heterochromatic regions by Swi6/HP1 in fission yeast". Nature Cell Biology. 4 (1): 89—93. January 2002. doi:10.1038/ncb739. PMID 11780129.
- ↑ "RNA interference machinery regulates chromosome dynamics during mitosis and meiosis in fission yeast". Proceedings of the National Academy of Sciences of the United States of America. 100 (1): 193—8. January 2003. Bibcode:2003PNAS..100..193H. doi:10.1073/pnas.232688099. PMID 12509501.
- ↑ "Dicer is essential for formation of the heterochromatin structure in vertebrate cells". Nature Cell Biology. 6 (8): 784—91. August 2004. doi:10.1038/ncb1155. PMID 15247924.
- ↑ "The kinetochore is an enhancer of pericentric cohesin binding". PLOS Biology. 2 (9): E260. September 2004. doi:10.1371/journal.pbio.0020260. PMID 15309047.
{{cite journal}}: Википедия:Обслуживание CS1 (не помеченный открытым DOI) (ссылка)
- ↑ "Human Orc2 localizes to centrosomes, centromeres and heterochromatin during chromosome inheritance". The EMBO Journal. 23 (13): 2651—63. July 2004. doi:10.1038/sj.emboj.7600255. PMID 15215892.
- ↑ "The origin recognition complex functions in sister-chromatid cohesion in Saccharomyces cerevisiae". Cell. 128 (1): 85—99. January 2007. doi:10.1016/j.cell.2006.11.045. PMID 17218257.
- ↑ "Kinetochore orientation in mitosis and meiosis". Cell. 119 (3): 317—27. October 2004. doi:10.1016/j.cell.2004.10.014. PMID 15507205.
- ↑ "Scc1/Rad21/Mcd1 is required for sister chromatid cohesion and kinetochore function in vertebrate cells". Developmental Cell. 1 (6): 759—70. December 2001. doi:10.1016/S1534-5807(01)00088-0. PMID 11740938.
- ↑ "Depletion of Drad21/Scc1 in Drosophila cells leads to instability of the cohesin complex and disruption of mitotic progression" (PDF). Current Biology. 13 (3): 208—18. February 2003. doi:10.1016/S0960-9822(03)00047-2. PMID 12573216. Архивировано (PDF) 20 сентября 2022. Дата обращения: 16 сентября 2022.
- ↑ "Molecular architecture of SMC proteins and the yeast cohesin complex". Molecular Cell. 9 (4): 773—88. April 2002. doi:10.1016/S1097-2765(02)00515-4. PMID 11983169.
- ↑ "SMC-mediated chromosome mechanics: a conserved scheme from bacteria to vertebrates?". Genes & Development. 13 (1): 11—9. January 1999. doi:10.1101/gad.13.1.11. PMID 9887095.
- ↑ "An ESP1/PDS1 complex regulates loss of sister chromatid cohesion at the metaphase to anaphase transition in yeast". Cell. 93 (6): 1067—76. June 1998. doi:10.1016/S0092-8674(00)81211-8. PMID 9635435.
- ↑ "Phosphorylation of the cohesin subunit Scc1 by Polo/Cdc5 kinase regulates sister chromatid separation in yeast". Cell. 105 (4): 459—72. May 2001. doi:10.1016/S0092-8674(01)00362-2. PMID 11371343.
- ↑ "Degradation of Drosophila PIM regulates sister chromatid separation during mitosis". Genes & Development. 14 (17): 2192—205. September 2000. doi:10.1101/gad.176700. PMID 10970883.
- ↑ "Securin degradation is mediated by fzy and fzr, and is required for complete chromatid separation but not for cytokinesis". The EMBO Journal. 20 (4): 792—801. February 2001. doi:10.1093/emboj/20.4.792. PMID 11179223.
- ↑ "Characterization of vertebrate cohesin complexes and their regulation in prophase". The Journal of Cell Biology. 151 (4): 749—62. November 2000. doi:10.1083/jcb.151.4.749. PMID 11076961.
- ↑ "Identification and characterization of SA/Scc3p subunits in the Xenopus and human cohesin complexes". The Journal of Cell Biology. 150 (3): 405—16. August 2000. doi:10.1083/jcb.150.3.405. PMID 10931856.
- ↑ "Regulation of sister chromatid cohesion between chromosome arms". Current Biology. 14 (13): 1187—93. July 2004. doi:10.1016/j.cub.2004.06.052. PMID 15242616.
- ↑ "Micromanipulation of chromosomes reveals that cohesion release during cell division is gradual and does not require tension". Current Biology. 14 (23): 2124—9. December 2004. doi:10.1016/j.cub.2004.11.052. PMID 15589155.
- ↑ "The complete removal of cohesin from chromosome arms depends on separase". Journal of Cell Science. 120 (Pt 23): 4188—96. December 2007. doi:10.1242/jcs.011528. PMID 18003702.
- ↑ "Shugoshin prevents dissociation of cohesin from centromeres during mitosis in vertebrate cells". PLOS Biology. 3 (3): e86. March 2005. doi:10.1371/journal.pbio.0030086. PMID 15737064.
{{cite journal}}: Википедия:Обслуживание CS1 (не помеченный открытым DOI) (ссылка) - ↑ "Vertebrate shugoshin links sister centromere cohesion and kinetochore microtubule stability in mitosis". Cell. 118 (5): 567—78. September 2004. doi:10.1016/j.cell.2004.08.016. PMID 15339662.
- ↑ 1 2 3 4 "The Mad1/Mad2 complex as a template for Mad2 activation in the spindle assembly checkpoint". Current Biology. 15 (3): 214—25. February 2005. doi:10.1016/j.cub.2005.01.038. PMID 15694304.
- ↑ 1 2 3 4 5 6 7 "The spindle-assembly checkpoint in space and time". Nature Reviews. Molecular Cell Biology. 8 (5): 379—93. May 2007. doi:10.1038/nrm2163. PMID 17426725.
- ↑ "Role of Hec1 in spindle checkpoint signaling and kinetochore recruitment of Mad1/Mad2". Science. 297 (5590): 2267—70. September 2002. Bibcode:2002Sci...297.2267M. doi:10.1126/science.1075596. PMID 12351790.
- ↑ "Survivin is required for a sustained spindle checkpoint arrest in response to lack of tension". The EMBO Journal. 22 (12): 2934—47. June 2003. doi:10.1093/emboj/cdg307. PMID 12805209.
- ↑ "The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint". The Journal of Cell Biology. 161 (2): 281—94. April 2003. doi:10.1083/jcb.200208092. PMID 12707311.
- ↑ "The kinetochore encodes a mechanical switch to disrupt spindle assembly checkpoint signalling". Nature Cell Biology. 17 (7): 868—79. July 2015. doi:10.1038/ncb3179. PMID 26053220.
- ↑ Bruce Alberts. Molecular biology of the cell. — Sixth edition. — New York, NY, 2015. — 1 volume (various pagings) с. — ISBN 978-0-8153-4432-2, 0-8153-4432-5, 978-0-8153-4464-3, 0-8153-4464-3, 978-0-8153-4524-4, 0-8153-4524-0. Архивировано 20 апреля 2021 года.
- ↑ "On the road to cancer: aneuploidy and the mitotic checkpoint". Nature Reviews. Cancer. 5 (10): 773—85. October 2005. doi:10.1038/nrc1714. PMID 16195750.
- ↑ "Genetic instabilities in human cancers". Nature. 396 (6712): 643—9. December 1998. Bibcode:1998Natur.396..643L. doi:10.1038/25292. PMID 9872311.
- ↑ "Does aneuploidy cause cancer?". Current Opinion in Cell Biology. 18 (6): 658—67. December 2006. doi:10.1016/j.ceb.2006.10.002. PMID 17046232.
- ↑ "Mutations of mitotic checkpoint genes in human cancers". Nature. 392 (6673): 300—3. March 1998. Bibcode:1998Natur.392..300C. doi:10.1038/32688. PMID 9521327.
- ↑ "Deficient spindle assembly checkpoint in multiple myeloma". PLOS ONE. 6 (11): e27583. 2011. Bibcode:2011PLoSO...627583D. doi:10.1371/journal.pone.0027583. PMID 22132115.
{{cite journal}}: Википедия:Обслуживание CS1 (не помеченный открытым DOI) (ссылка) - ↑ Grady, William M. (2004). "Genomic instability and colon cancer". Cancer and Metastasis Reviews. 23 (1—2): 11—27. doi:10.1023/A:1025861527711. PMID 15000146.
- ↑ "A requirement for breast-cancer-associated gene 1 (BRCA1) in the spindle checkpoint". Proceedings of the National Academy of Sciences of the United States of America. 101 (49): 17108—13. December 2004. Bibcode:2004PNAS..10117108W. doi:10.1073/pnas.0407585101. PMID 15563594.
- ↑ "Mad2 overexpression promotes aneuploidy and tumorigenesis in mice". Cancer Cell. 11 (1): 9—23. January 2007. doi:10.1016/j.ccr.2006.10.019. PMID 17189715.
- ↑ "Overexpression of BUBR1 is associated with chromosomal instability in bladder cancer". Cancer Genetics and Cytogenetics. 174 (1): 42—7. April 2007. doi:10.1016/j.cancergencyto.2006.11.012. PMID 17350465.
- ↑ 1 2 "The role of aneuploidy in promoting and suppressing tumors". The Journal of Cell Biology. 185 (6): 935—7. June 2009. doi:10.1083/jcb.200905098. PMID 19528293.
- ↑ Cross, Shawn M. (1995). "A p53-dependant mouse spindle checkpoint". Science. 3 (5202): 1353—1356. Bibcode:1995Sci...267.1353C. doi:10.1126/science.7871434. PMID 7871434.
- ↑ "The molecular basis and potential role of survivin in cancer diagnosis and therapy". Trends in Molecular Medicine. 7 (12): 542—7. December 2001. doi:10.1016/S1471-4914(01)02243-2. PMID 11733216.
- ↑ "When the genome plays dice: circumvention of the spindle assembly checkpoint and near-random chromosome segregation in multipolar cancer cell mitoses". PLOS ONE. 3 (4): e1871. April 2008. Bibcode:2008PLoSO...3.1871G. doi:10.1371/journal.pone.0001871. PMID 18392149.
{{cite journal}}: Википедия:Обслуживание CS1 (не помеченный открытым DOI) (ссылка) - ↑ "Targeting microtubules for cancer chemotherapy". Current Medicinal Chemistry. Anti-Cancer Agents. 5 (1): 65—71. January 2005. doi:10.2174/1568011053352569. PMID 15720262.
- ↑ 1 2 "Aurora kinases: new targets for cancer therapy". Clinical Cancer Research. 12 (23): 6869—75. December 2006. doi:10.1158/1078-0432.CCR-06-1405. PMID 17145803.
- ↑ "AURORA-A amplification overrides the mitotic spindle assembly checkpoint, inducing resistance to Taxol". Cancer Cell. 3 (1): 51—62. January 2003. doi:10.1016/S1535-6108(02)00235-0. PMID 12559175.
- ↑ "VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo". Nature Medicine. 10 (3): 262—7. March 2004. doi:10.1038/nm1003. PMID 14981513.
- ↑ "Survivin, cancer networks and pathway-directed drug discovery". Nature Reviews. Cancer. 8 (1): 61—70. January 2008. doi:10.1038/nrc2293. PMID 18075512.
- ↑ "Induction of apoptosis by an inhibitor of the mitotic kinesin KSP requires both activation of the spindle assembly checkpoint and mitotic slippage". Cancer Cell. 8 (1): 49—59. July 2005. doi:10.1016/j.ccr.2005.06.003. PMID 16023598.
Дальнейшее чтение
- Larsen NA, Al-Bassam J, Wei RR, Harrison SC (January 2007). "Structural analysis of Bub3 interactions in the mitotic spindle checkpoint". Proceedings of the National Academy of Sciences of the United States of America. 104 (4): 1201—6. Bibcode:2007PNAS..104.1201L. doi:10.1073/pnas.0610358104. PMC 1770893. PMID 17227844.
- Wang X, Babu JR, Harden JM, Jablonski SA, Gazi MH, Lingle WL, de Groen PC, Yen TJ, van Deursen JM (July 2001). "The mitotic checkpoint protein hBUB3 and the mRNA export factor hRAE1 interact with GLE2p-binding sequence (GLEBS)-containing proteins". The Journal of Biological Chemistry. 276 (28): 26559—67. doi:10.1074/jbc.M101083200. PMID 11352911.
- Kitagawa R, Rose AM (December 1999). "Components of the spindle-assembly checkpoint are essential in Caenorhabditis elegans". Nature Cell Biology. 1 (8): 514—21. doi:10.1038/70309. PMID 10587648. S2CID 25953096.
Ссылки
- Лаборатория Теда Сэлмона: фильмы о делящихся клетках. [1]
- Лаборатория Андреа Мусаккио: схемы контрольных точек шпинделя. [2]
- http://www.uniprot.org/uniprot/O60566
| Фазы |
|  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Регуляторы |
| ||||||||||