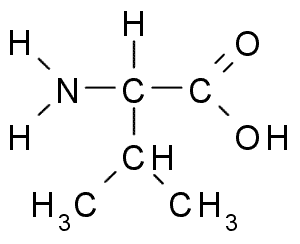
Вали́н — алифатическая α-аминокислота, одна из 20 протеиногенных аминокислот, входит в состав практически всех известных белков. Названо в честь растения валерианы.

Лейцин (сокр. Leu или L; 2-амино-4-метилпентановая кислота; от греч. leukos — «белый») — алифатическая аминокислота с химической формулой HO2CCH(NH2)CH2CH(CH3)2. Имеет в своей структуре один хиральный центр и может существовать в виде D- или L-оптических изомера, а также в виде рацемата (смеси равных количеств D- и L-изомера). В живых организмах встречается в виде L-изомера.

Фенилкетонури́я — наследственное заболевание группы ферментопатий, связанное с нарушением метаболизма аминокислот, главным образом фенилаланина. Несоблюдение низкобелковой диеты сопровождается накоплением фенилаланина и его токсических продуктов, что приводит к тяжёлому поражению ЦНС, проявляющемуся, в частности, в виде нарушения умственного развития. Одно из немногих наследственных заболеваний, поддающихся успешному лечению.

Муковисцидóз — системное наследственное заболевание, обусловленное мутацией гена трансмембранного регулятора муковисцидоза и характеризующееся поражением желёз внешней секреции, тяжёлыми нарушениями функций органов дыхания. Муковисцидоз представляет особый интерес не только из-за широкой распространённости, но и потому, что это одно из первых наследственных заболеваний, которое пытались лечить. Впервые муковисцидоз был признан отдельной нозологией Дороти Андерсен в 1938 году.

Общий ана́лиз мочи́ — лабораторное исследование мочи, проводимое для нужд медицинской практики, как правило, с диагностической целью. Включает органолептическое, физико-химическое и биохимическое исследования, а также микробиологическое исследование и микроскопическое изучение мочевого осадка.
Почечная недостаточность — синдром нарушения всех функций почек, приводящего к расстройству водного, электролитного, азотистого и других видов обмена. Различают острую и хроническую почечную недостаточность.
Массовое обследование новорождённых — один из эффективных способов выявления наиболее распространенных врождённых и наследственных заболеваний у новорождённых детей.
Синдро́м (болезнь) де То́ни — Дебре́ — Фанко́ни — врождённое заболевание, наследуется по аутосомно-рецессивному типу. Комплекс биохимических и клинических проявлений поражения проксимальных почечных канальцев с нарушением канальцевой реабсорбции фосфата, глюкозы, аминокислот и бикарбоната. Одно из рахитоподобных заболеваний.
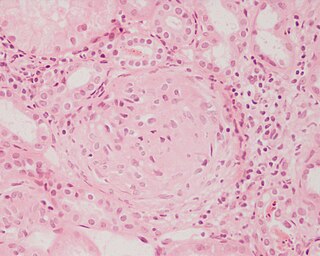
Гломерулонефри́т — заболевание почек, характеризующееся поражением гломерул. Это состояние может быть представлено изолированной гематурией и/или протеинурией; или как нефритический синдром, острая почечная недостаточность, или хроническая почечная недостаточность. Они собраны в несколько различных групп — непролиферативные или пролиферативные типы. Диагностирование образца ГН важно, потому что тактика и лечение отличаются в зависимости от типа.
6-я хромосо́ма челове́ка — одна из 24 человеческих хромосом. Хромосома содержит более 171 млн пар оснований, что составляет от 5,5 до 6 % всего материала ДНК человеческой клетки. В настоящее время считается, что на 6-й хромосоме находятся от 1100 до 1600 генов.
Генные болезни — это большая группа заболеваний, возникающих в результате повреждения ДНК на уровне гена. Термин употребляется в отношении моногенных заболеваний, в отличие от более широкой группы — Наследственные заболевания (см.)

Гипогликеми́ческий синдро́м — клинический симптомокомплекс, развивающийся вследствие дисбаланса в системе регуляции уровня глюкозы в плазме крови, приводящий к гипогликемической реакции организма. Гипогликемией принято считать снижение плазменной концентрации глюкозы до уровня ниже 2,2—2,8 ммоль/л. Наиболее часто гипогликемии развиваются в процессе лечения сахарного диабета, однако эти состояния не включены в понятие гликемический синдром.

Аминокислоты с разветвлёнными боковыми цепями — группа протеиногенных аминокислот, характеризующихся разветвлёнными строением алифатической боковой цепи. К таким аминокислотам относятся лейцин, изолейцин и валин.
Паренхимато́зные диспротеино́зы — дисметаболические процессы с преимущественным нарушением обмена белков, развивающиеся первично в паренхиматозных клетках органов.
Первичные иммунодефициты — наследственные или приобретённые во внутриутробном периоде иммунодефицитные состояния. Обычно они проявляются или сразу после рождения, или в течение первых двух лет жизни. Однако менее выраженные генетические дефекты иммунного ответа могут манифестировать позже, например, на втором—третьем десятилетии жизни. Наследственные формы первичных иммунодефицитов, как правило, характеризуются аутосомно-рецессивным или рецессивным, сцепленным с Х-хромосомой типом наследования. Первым в 1952 г. был описан синдром Бру́тона.
Пропионовая ацидемия — это органическая ацидемия, вызванная мутациями в генах PCCA и PCCB. Расстройство проявляется в раннем неонатальном периоде плохим питанием, рвотой, вялостью и отсутствием мышечного тонуса, без лечения смерть может наступить быстро, из-за вторичной гипераммонемии, кетоацидоза, или повреждения головного мозга. Особая разновидность пропионовой ацидемии может проявиться в более поздном возрасте в виде кардиопатии без кетоацидоза. В некоторых странах, пропионовая ацидемия включена в неонатальный скриниг, что позволяет определять больных до наступления симптомов и ускорять постановку правильного диагноза. Биохимически, заболевание можно подтвердить наличием метилмалоновой кислоты в крови или моче и повышенным пропионилкарнитином (С3) в плазме крови.
Дегидрогеназный комплекс разветвлённых α-кетокислот, также дегидрогеназа α-кетокислот с разветвлённой цепью — мультиферментный комплекс, локализованный на внутренней мембране митохондрий. Данный комплекс катализирует реакцию окислительного декарбоксилирования разветвлённых α-кетокислот с короткой углеродной цепью. Дегидрогеназный комплекс структурно аналогичен пируватдегидрогеназному и α-кетоглутаратдегидрогеназному комплексам, образуя вместе с ними семейство митохондриальных ферментов, участвующих в окислительном декарбоксилировании α-кетокислот.
Недостаточность киназы дегидрогеназы α-кетокислот с разветвлённой цепью — заболевание, при котором избыточная активность дегидрогеназного комплекса приводит к недостаточности аминокислот с разветвлёнными боковыми цепями. У пациентов отмечается задержка в развитии, признаки аутизма, развиваются эпилептические приступы.
Комбинированная малоновая и метилмалоновая ацидурия (КМАММА), также называемая комбинированной малоновой и метилмалоновой ацидемией — это наследственное метаболическое заболевание, характеризующееся повышенным уровнем малоновой кислот и метилмалоновой кислот. Некоторые исследователи предположили, что КМАММА может быть одной из самых распространенных форм метилмалоновой ацидемии и, возможно, одной из самых распространенных врожденных ошибок метаболизма. Из-за того, что диагноз ставится нечасто, чаще всего он остается невыявленным.

Киназа дегидрогеназы α-кетокислот с разветвлённой цепью — фермент из семейства протеинкиназ, катализирующий реакцию фосфорилирования или переноса фосфатной группы от молекулы ATP, на остаток серина дегидрогеназы α-кетокислот с разветвлённой цепью (BCKDC). Этот фермент является частью семейства митохондриальных протеинкиназ и регулятором катаболических путей разветвлённых аминокислот (BCAA) — валина, лейцина и изолейцина. BCKDK обнаруживается в митохондриальном матриксе, и его распространённость зависит от типа клетки. Гепатоциты, как правило, имеют самую низкую концентрацию BCKDK, тогда как клетки скелетных мышц имеют самую высокую концентрацию. Аномальная активность этого фермента часто приводит к таким заболеваниям, как лейциноз и кахексия.