Лизосома
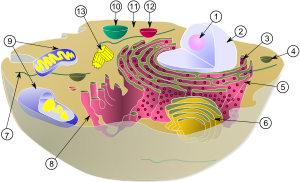
(1) Ядрышко
(2) Ядро
(3) Рибосома (маленькие точки)
(4) Везикула
(5) Шероховатый эндоплазматический ретикулум (ER)
(6) Аппарат Гольджи
(7) Цитоскелет
(8) Гладкий эндоплазматический ретикулум
(9) Митохондрия
(10) Вакуоль
(11) Цитоплазма
(12) Лизосома
(13) Центриоль и Центросома

Лизосо́ма (от греч. λύσις — разложение и σώμα — тело) — окружённая мембраной клеточная органелла, в полости которой поддерживается кислая среда и находится множество растворимых гидролитических ферментов[1]. Лизосома отвечает за внутриклеточное переваривание макромолекул, в том числе при аутофагии; лизосома способна к секреции своего содержимого во время экзоцитоза; также лизосома участвует в некоторых внутриклеточных сигнальных путях, связанных с метаболизмом и ростом клетки[2].
Лизосома является одним из видов везикул и относится к эндомембранной системе клетки[3]. Разные виды лизосом могут рассматриваться как отдельные клеточные компартменты.
Лизосомы были открыты в 1955 году бельгийским биохимиком Кристианом де Дювом[4]. Лизосомы есть во всех клетках млекопитающих, за исключением эритроцитов[5]. У растений к лизосомам по способу образования, а отчасти и по функциям близки вакуоли. Лизосомы есть также у большинства протистов (как с фаготрофным, так и с осмотрофным типом питания) и у грибов. Таким образом, наличие лизосом характерно для клеток всех эукариот. У прокариот лизосомы отсутствуют, так как у них отсутствует фагоцитоз и нет внутриклеточного пищеварения.
С нарушением функций лизосом связан ряд наследственных заболеваний у человека, называемых лизосомными болезнями накопления[6].
История открытия
В 1949—1952 годах биохимик Кристиан де Дюв и его студенты, изучавшие действие инсулина в клетках печени крыс, случайно обнаружили неожиданное различие в активности кислой фосфатазы в зависимости от способа выделения. Кислая фосфатаза использовалась ими в качестве стандарта, основным предметом их изучения был фермент глюкозо-6-фосфатаза, вовлечённый в метаболизм инсулина. В ходе экспериментов выяснилось, что при фракционировании клеточного содержимого на центрифуге кислая фосфатаза была ассоциирована с микросомальной фракцией, но проявляла только десятую часть активности в сравнении с простым клеточным экстрактом, причём после нескольких дней хранения микросомальной фракции в холодильнике активность кислой фосфатазы возрастала. При обнаружении этого феномена первым объяснением было то, что произошла какая-то техническая ошибка. Однако повторение эксперимента неизменно воспроизводило первоначальную картину. Это позволило предположить существование неких окружённых мембраной клеточных частиц, которые содержат внутри себя фермент. С 1952 по 1955 год было открыто ещё несколько кислых гидролаз, связанных с микросомальной фракцией. В 1955 году, который считается годом открытия лизосом, К. де Дюв предложил название «лизосома» для клеточной органеллы, которая окружена мембраной, внутри которой поддерживается низкий pH и внутри которой находится ряд ферментов, оптимально работающих в кислой среде[7][8]. В том же 1955 году американский цитолог Алекс Новиков[англ.] из Вермонтского университета США, блестяще владевший техникой микроскопии, посетил лабораторию К. де Дюве и смог получить первые электронные фотографии этих органелл, используя препарат частично очищенных лизосом. Позднее в 1961 году Алекс Новиков с помощью гистохимического выявления кислой фосфатазы и электронной микроскопии подтвердил локализацию этого фермента в лизосомах[9][10]. В 1963 году бельгийский биохимик Анри Эрс, ранее работавший в группе К. де Дюве, обнаружил недостаточность лизосомного фермента α-глюкозидазы у пациентов с болезнью Помпе и высказал предположение о связи других генетических заболеваний с нарушением работы лизосом[11]. В настоящее время более 50 наследственных заболеваний связывают с лизосомной недостаточностью[12].
В 1974 году за свой вклад в раскрытие структурной и функциональной организации клетки К. де Дюв был удостоен Нобелевской премии по медицине[13].
Признаки лизосом
Лизосомы являются гетерогенными по форме, размеру, ультраструктурным и цитохимическим особенностям. В клетках животных размер лизосом составляет обычно менее 1 мкм, хотя в некоторых типах клеток, например, в макрофагах, размер лизосом может превышать несколько микрон. Лизосомы, как правило, имеют сферическую, овальную, иногда тубулярную форму[14]. Число лизосом варьирует от одной (крупная вакуоль во многих клетках растений и грибов) до нескольких сотен или тысяч (в клетках животных). Лизосомы у животных обычно составляют не более 5 % внутриклеточного объёма[15].
Один из признаков лизосом — наличие в них ряда ферментов (кислых гидролаз), способных расщеплять белки, углеводы, липиды и нуклеиновые кислоты. К числу ферментов лизосом относятся катепсины (тканевые протеазы), кислая рибонуклеаза, фосфолипаза и др. Кроме того, в лизосомах присутствуют ферменты, которые способны отщеплять от органических молекул сульфатные (сульфатазы) или фосфатные (кислая фосфатаза) группы. Всего полость лизосомы содержит около 60 растворимых кислых гидролитических ферментов[2].
Для лизосом характерна кислая реакция внутренней среды, которая обеспечивает оптимум работы лизосомных гидролаз[14]. Обычно pH в лизосомах составляет около 4,5-5, то есть концентрация протонов в них на два порядка выше, чем в цитоплазме. Это обеспечивается активным транспортом протонов, который осуществляет встроенный в мембраны лизосом белок-насос протонная АТФаза[15]. Помимо протонного насоса в мембрану лизосом встроены белки-переносчики для транспорта в цитоплазму продуктов гидролиза макромолекул: аминокислот, сахаров, нуклеотидов, липидов[16].
Высокая активность кислой фосфатазы ранее использовалась как один из маркеров лизосом. В настоящее время более надежным маркером считается присутствие специфических мембранных гликопротеидов — LAMP1 и LAMP2. Они присутствуют на мембране лизосом и поздних эндосом, но отсутствуют на мембранах других компартментов вакуома.
Образование лизосом и их типы
Лизосомы формируются из пузырьков (везикул), отделяющихся от аппарата Гольджи, и пузырьков (эндосом), в которые попадают вещества при эндоцитозе[17]. В образовании аутолизосом (аутофагосом) принимают участие мембраны эндоплазматического ретикулума. Все белки лизосом синтезируются на «сидячих» рибосомах на внешней стороне мембран эндоплазматического ретикулума и затем проходят через его полость и через аппарат Гольджи.
Общепринятой классификации и номенклатуры для разных стадий созревания и типов лизосом нет. Различают первичные и вторичные лизосомы. Первые образуются в области аппарата Гольджи, в них находятся ферменты в неактивном состоянии, вторые же содержат активные ферменты. Обычно ферменты лизосом активируются при понижении рН. Среди лизосом можно также выделить гетеролизосомы (переваривающие материал, поступающий в клетку извне — путём фаго- или пиноцитоза) и аутолизосомы (разрушающие собственные белки или органеллы клетки). Наиболее широко используется следующая классификация лизосом и связанных с ними компартментов:
- Ранняя эндосома — в неё поступают эндоцитозные (пиноцитозные) пузырьки. Из ранней эндосомы рецепторы, отдавшие (из-за пониженного рН) свой груз, возвращаются на наружную мембрану.
- Поздняя эндосома — в неё из ранней эндосомы поступают пузырьки с материалом, поглощённом при пиноцитозе, и пузырьки из аппарата Гольджи с гидролазами. Рецепторы маннозо-6-фосфата возвращаются из поздней эндосомы в аппарат Гольджи.
- Лизосома — в неё из поздней эндосомы поступают пузырьки со смесью гидролаз и перевариваемого материала.
- Фагосома — в неё попадают более крупные частицы (бактерии и т. п.), поглощённые путём фагоцитоза. Фагосомы обычно сливаются с лизосомой.
- Аутофагосома — окружённый двумя мембранами участок цитоплазмы, обычно включающий какие-либо органеллы и образующийся при макроаутофагии. Сливается с лизосомой.
- Мультивезикулярные тельца — обычно окружены одинарной мембраной, содержат внутри более мелкие окружённые одинарной мембраной пузырьки. Образуются в результате процесса, напоминающего микроаутофагию (см. ниже), но содержат материал, полученный извне. В мелких пузырьках обычно остаются и затем подвергаются деградации рецепторы наружной мембраны (например, рецепторы эпидермального фактора роста). По стадии формирования соответствуют ранней эндосоме. Описано образование мультивезикулярных телец, окруженных двумя мембранами, путём отпочковывания от ядерной оболочки.
- Остаточные тельца (телолизосомы) — пузырьки, содержащие непереваренный материал (в частности, липофусцин). В нормальных клетках сливаются с наружной мембраной и путём экзоцитоза покидают клетку. При старении или патологии накапливаются.
Функции лизосом
Функциями лизосом являются:
- переваривание захваченных клеткой при эндоцитозе веществ или частиц (бактерий, других клеток).
- аутофагия — уничтожение ненужных клетке структур, к примеру, во время замены старых органелл новыми, или переваривание белков и других веществ, произведенных внутри самой клетки.
- автолиз — самопереваривание клетки, приводящее к её гибели (иногда этот процесс не является патологическим, а сопровождает развитие организма или дифференцировку некоторых специализированных клеток). Пример: При превращении головастика в лягушку, лизосомы, находящиеся в клетках хвоста, переваривают его: хвост исчезает, а образовавшиеся во время этого процесса вещества всасываются и используются другими клетками тела.
- растворение внешних структур (см, например, остеокласты)
Внутриклеточное пищеварение и участие в обмене веществ
У многих протистов и у животных, имеющих внутриклеточное пищеварение, лизосомы участвуют в переваривании пищи, захваченной путём эндоцитоза. При этом лизосомы сливаются с пищеварительными вакуолями. У протистов непереваренные остатки пищи обычно удаляются из клетки при слиянии пищеварительной вакуоли с наружной мембраной.
Многие клетки животных, у которых преобладает полостное пищеварение (например, хордовые) получают питательные вещества из межклеточной жидкости или плазмы крови с помощью пиноцитоза. Эти вещества также вовлекаются в обмен веществ клетки после их переваривания в лизосомах. Хорошо изученный пример такого участия лизосом в обмене веществ — получение клетками холестерина. Холестерин, приносимый кровью в виде ЛПНП, поступает внутрь пиноцитозных везикул после соединения ЛПНП с рецепторами ЛПНП на мембране. Рецепторы возвращаются к мембране из ранней эндосомы, а ЛПНП поступают в лизосомы. После этого ЛПНП перевариваются, а высвободившийся холестерин через мембрану лизосом поступает в цитоплазму.
Косвенно лизосомы участвуют в обмене, обеспечивая десенсибилизацию клеток к воздействию гормонов. При длительном действии гормона на клетку часть рецепторов, связавших гормон, поступают в эндосомы и затем деградируют внутри лизосом. Снижение числа рецепторов понижает чувствительность клетки к гормону.
Аутофагия
Обычно различают два типа аутофагии — микроаутофагия и макроаутофагия. При микроаутофагии, как при образовании мультивезикулярных телец, образуются впячивания мембраны эндосомы или лизосомы, которые затем отделяются в виде внутренних пузырьков, только в них попадают вещества, синтезированные в самой клетке. Таким путём клетка может переваривать белки при нехватке энергии или строительного материала (например, при голодании). Но процессы микроаутофагии происходят и при нормальных условиях и в целом неизбирательны. Иногда в ходе микроаутофагии перевариваются и органеллы; так, у дрожжей описана микроаутофагия пероксисом и частичная микроаутофагия ядер, при которой клетка сохраняет жизнеспособность.
При макроаутофагии участок цитоплазмы (часто содержащий какие-либо органеллы) окружается мембранным компартментом, похожим на цистерну эндоплазматической сети. В результате этот участок оказывается отгорожен от остальной цитоплазмы двумя мембранами. Затем такая аутофагосома сливается с лизосомой, и её содержимое переваривается. Видимо, макроаутофагия также неизбирательна, хотя часто подчеркивается, что с помощью неё клетка может избавляться от «отслуживших свой срок» органелл (митохондрий, рибосом и др.).
Третий тип аутофагии — шаперон-зависимая. При этом способе происходит направленный транспорт частично денатурировавших белков из цитоплазмы сквозь мембрану лизосомы в её полость.
Автолиз
Ферменты лизосом нередко высвобождаются при разрушении мембраны лизосомы. Обычно при этом они инактивируются в нейтральной среде цитоплазмы. Однако при одновременном разрушении всех лизосом клетки может произойти её саморазрушение — автолиз. Различают патологический и обычный автолиз. Распространенный вариант патологического автолиза — посмертный автолиз тканей.
В норме процессы автолиза сопровождают многие явления, связанные с развитием организма и дифференцировкой клеток. Так, автолиз клеток описывается как механизм разрушения тканей у личинок насекомых при полном превращении, а также при рассасывании хвоста у головастика. Правда, эти описания относятся к периоду, когда различия между апоптозом и некрозом ещё не были установлены, и в каждом случае требуется выяснять, не лежит ли на самом деле в основе деградации органа или ткани апоптоз, не связанный с автолизом.
У растений автолизом сопровождается дифференциация клеток, которые функционируют после смерти (например, трахеид или члеников сосудов). Частичный автолиз происходит и при созревании клеток флоэмы- члеников ситовидных трубок.
Клиническое значение
Иногда из-за неправильной работы лизосом развиваются болезни накопления, при которых ферменты из-за мутаций не работают или работают плохо. Примером лизосомных болезней накопления может служить болезнь Гоше, болезнь Помпе, Болезнь Тея — Сакса. Всего известно более 50 наследственных заболеваний, связанных с нарушением функции лизосомы[12].
Повреждение лизосом некротизированных клеток, в том числе гранулоцитов, даёт начало воспалительному процессу[18].
См. также
Примечания
- ↑ Альбертс и др., 2013, с. 1196.
- ↑ 1 2 Settembre C. et al. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. (англ.) // Nat. Rev. Mol. Cell Biol. — 2013. — Vol. 14. — P. 283-296. — doi:10.1038/nrm3565.
- ↑ Brighouse A., Dacks J. B., Field M. C. Rab protein evolution and the history of the eukaryotic endomembrane system (англ.) // Cellular and molecular life sciences. — 2010. — Vol. 67, no. 20. — P. 3449-3465. — doi:10.1007/s00018-010-0436-1. Архивировано 6 января 2015 года.
- ↑ De Duve C. The lysosome turns fifty (англ.) // Nature cell biology. — 2005. — Vol. 7, no. 9. — P. 847-849.
- ↑ Lüllmann-Rauch R. History and morphology of the lysosome // Lysosomes / P. Saftig. — Springer US, 2005. — P. 1-16. — ISBN 978-0-387-25562-0.
- ↑ Platt F. M., Boland B., van der Spoel A. C. Lysosomal storage disorders: The cellular impact of lysosomal dysfunction (англ.) // The Journal of cell biology. — 2012. — Vol. 199, no. 5. — P. 723-734. — doi:10.1083/jcb.201208152. Архивировано 1 января 2015 года.
- ↑ De Duve C. et al. Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue (англ.) // Biochemical Journal. — 1955. — Vol. 60, no. 4. — P. 604–617. Архивировано 22 января 2022 года.
- ↑ Bainton D. F. The discovery of lysosomes (англ.). — 1981. — Vol. 91. — P. 66-76. Архивировано 24 декабря 2012 года.
- ↑ Essner E., Novikoff A. B. Localization of acid phosphatase activity in hepatic lysosomes by means of electron microscopy (англ.) // The Journal of biophysical and biochemical cytology. — 1961. — Vol. 9, no. 4. — P. 773-784.
- ↑ Castro-Obregon S. The Discovery of Lysosomes and Autophagy (англ.) // Nature Education. — 2010. — Vol. 3, no. 9. — P. 49. Архивировано 23 декабря 2014 года.
- ↑ Hers H. G. α-Glucosidase deficiency in generalized glycogen-storage disease (Pompe's disease) (англ.) // Biochemical Journal. — 1963. — Vol. 86, no. 1. — P. 11-16. — PMID 13954110. Архивировано 21 января 2022 года.
- ↑ 1 2 la Marca G. Lysosomals // Physician's Guide to the Diagnosis, Treatment, and Follow-Up of Inherited Metabolic Diseases / N. Blau, M. Duran, K. M. Gibson, C. D. Vici. — Springer Berlin Heidelberg, 2014. — P. 785-793. — ISBN 978-3-642-40336-1.
- ↑ The Nobel Prize in Physiology or Medicine 1974 (англ.). Nobelprize.org. Nobel Media AB 2014. Дата обращения: 3 января 2015. Архивировано 15 октября 2012 года.
- ↑ 1 2 Appelqvist H. et al. The lysosome: from waste bag to potential therapeutic target (англ.) // Journal of molecular cell biology. — 2013. — Vol. 5, no. 4. — P. 214-226. — doi:10.1093/jmcb/mjt022. Архивировано 12 августа 2015 года.
- ↑ 1 2 Luzio J. P., Pryor P. R., Bright N. A. Lysosomes: fusion and function (англ.) // Nat. Rev. Mol. Cell Biol. — 2007. — Vol. 8. — P. 622-632. — doi:10.1038/nrm2217.
- ↑ Ченцов Ю. С. Введение в клеточную биологию. Учебник для вузов / Ю. С. Ченцов. — 4-e. — М.: ИКЦ «Академкнига», 2004. — 495 с.
- ↑ Saftig P., Klumperman J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function // Nat. Rev. Mol. Cell Biol. — 2009. — Vol. 10. — P. 623-635. — doi:10.1038/nrm2745. Архивировано 24 декабря 2012 года.
- ↑ Тель Л.З., Лысенков С.П., Шарипова Н.Г., Шастун С.А. Патофизиология и физиология в вопросах и ответах. — 2 том. — М.: Медицинское информационное агентство, 2007. — С. 67. — 512 с.
Литература
- Ченцов Ю. С. Цитология с элементами целлюлярной патологии: Учебное пособие для университетов и медицинских вузов. — М.: МИА, 2010. — 361 с. — 4000 экз. — ISBN 978-5-9986-0013-5.
- Молекулярная биология клетки: в 3-х томах / Б. Альбертс, А. Джонсон, Д. Льюис и др. — М.-Ижевск: НИЦ «Регулярная и хаотическая динамика», Институт компьютерных исследований, 2013. — Т. II. — С. 1196-1208. — 992 с. — ISBN 978-5-4344-0112-8.
- Клетки / Б. Льюин и др. — М.: БИНОМ. Лаборатория знаний, 2011. — С. 179-235. — 951 с. — (Лучший зарубежный учебник). — ISBN 978-5-94774-794-2.
| Словари и энциклопедии | |
|---|---|
| В библиографических каталогах |
| Эндомембранная система | |
|---|---|
| Цитоскелет | |
| Эндосимбионты | |
| Другие внутренние органеллы | |
| Внешние органеллы | |












