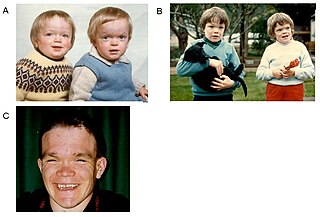
Лизосомные болезни накопления
| Лизосомные болезни накопления | |
|---|---|
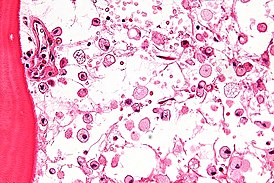 На микрофотографии характерные изменения строения костного мозга при болезни Гоше — цитоплазма макрофагов напоминает смятую папиросную бумагу. Окраска гематоксилином и эозином | |
| МКБ-11 | 5C56 |
| МКБ-10 | E75-E77 |
| MeSH | D016464 |
Лизосо́мные боле́зни накопле́ния (англ. Lysosomal Storage Diseases) — общее название группы весьма редких наследственных заболеваний, вызванных нарушением функции внутриклеточных органелл лизосом. Эти одномембранные органоиды являются частью эндомембранной системы клетки и специализируются на внутриклеточном расщеплении веществ: гликогена, гликозаминогликанов, гликопротеинов и других. Лизосомные болезни накопления вызываются генетически обусловленным дефицитом ферментов лизосом, что приводит к накоплению макромолекул, являющихся субстратом этих ферментов, в различных органах и тканях организма[1][2][3][4][5][6].
Данная группа объединяет мукополисахаридозы, муколипидозы, гликогенозы, болезни накопления липидов, гликопротеинов и других макромолекул.
Историческая справка
Клиническая картина первого наследственного заболевания из группы лизосомных болезней накопления (болезнь Тея — Сакса) была описана в 1881 году[7].
Затем, в 1882 году[8] описано заболевание, названное в честь впервые описавшего его французского врача Филиппа Гоше.
В 1932 году голландский врач Иоанн Помпе описал гликогеноз второго типа, впоследствии названный по его имени болезнью Помпе[9].
В конце 1950-х — начале 1960-х годов бельгийский биохимик Кристиан де Дюв с соавторами, используя методику фракционирования клеток, открыл лизосомы в качестве клеточных органелл[10][11][12][13][14][15][16][17][18][19][20][21][22], ответственных за расщепление и утилизацию макромолекул. Данное научное открытие дало возможность вскоре выявить патофизиологическую основу лизосомных болезней накопления[23].
Болезнь Помпе стала первым наследственным заболеванием, идентифицированным как лизосомная болезнь накопления. В 1963 году бельгийский физиолог и биохимик Анри Эрс (англ. Henri G. Hers) опубликовал работу, в которой связал причину развития данного симптомокомплекса с дефицитом α-глюкозидазы и высказал предположение о связи других генетических заболеваний, в том числе мукополисахаридозов, с недостаточностью того или иного фермента[24].
Эпидемиология
По состоянию на 2014 год, известно свыше 50 лизосомных болезней накопления, среди 7000—8000 новорождённых диагностируют около одного заболевания этой группы[25][26]. Каждое заболевание индивидуально встречается не чаще, чем у одного из 100 тысяч новорождённых, при этом распространённость конкретного заболевания в различных популяциях может значительно варьировать[25]. Распространение мутаций, ведущих к какому-либо заболеванию из группы лизосомных болезней накопления, определяется популяционно-генетическими факторами и, в первую очередь, связано с эффектом основателя. Типичным следствием эффекта основателя являются различие по наиболее распространённым мутациям одного и того же гена в различных генетически изолированных популяциях и этнических группах[27].
Наследование
Подавляющее большинство лизосомных болезней накопления наследуются аутосомно-рецессивно, за исключением трёх заболеваний, которые наследуются сцепленно с полом. К этим трём заболеваниям относятся мукополисахаридоз Хантера (МПС II) и болезнь Фабри, являющиеся рецессивными Х-сцепленными заболеваниями, а также синдром Данона[англ.][28], являющийся доминантным Х-сцепленным заболеванием[2][3][5][25].
Патогенез
Большая часть заболеваний группы связана с генетически обусловленным нарушением функции какой-либо из лизосомных гидролаз. Это приводит к прогрессивному накоплению внутри клетки промежуточного субстрата, который в норме распадается[28]. Некоторые болезни этой группы вызваны нарушениями белков, участвующих в везикулярном транспорте или в биогенезе лизосом. Например, муколипидоз II типа (I-клеточная болезнь) вызван дефектом фосфоэстеразы, которая локализована в аппарате Гольджи, нарушение работы этого фермента приводит к ошибочной переадресации лизосомных гидролаз во внеклеточное пространство вместо их транспорта из аппарата Гольджи в лизосому[29]. Хотя ферменты лизосом относятся к белкам, которые экспрессируются в подавляющем большинстве типов клеток, аномальное накопление субстратных макромолекул происходит в тех клетках, тканях и органах, которые характеризуются повышенной скоростью обновления этих макромолекул. Первичное накопление макромолекул может вести к вторичным нарушениям других биохимических и клеточных функций, что обуславливает обычно тяжёлое течение заболеваний этой группы[30].
Классификация
В связи с тем, что при многих лизосомных болезнях накопления наблюдается сходная клиническая картина, их классифицируют в соответствии с типом вещества, которое накапливается (например, мукополисахаридозы, гликопротеинозы, сфинголипидозы)[28].
| Мукополисахаридозы (MPS) |
|
|---|---|
| Муколипидозы (ML) |
|
| Сфинголипидозы | |
| Олигосахаридозы |
|
| Восковидныелипофусцинозынейронов | |
| Прочие | |
Ниже приведена классификация, согласно действующей МКБ (указан код МКБ-10):
- E72 Другие нарушения обмена аминокислот.
- E74 Другие нарушения обмена углеводов.
- E74.0 Болезни накопления гликогена. Болезнь Помпе.
- E75 Нарушения обмена сфинголипидов и другие болезни накопления липидов.
- E75.0 GM2 ганглиозидоз: Болезнь Сандхоффа, Болезнь Тея — Сакса, GM2-ганглиозидоз: БДУ (без дополнительных уточнений), взрослых, ювенильный.
- E75.1 Другие ганглиозидозы. Ганглиозидоз GM1, Муколипидоз IV.
- E75.2 Другие сфинголипидозы. Болезнь Фабри. Болезнь Гоше. Болезнь Краббе. Болезнь Ниманна — Пика. Синдром Фарбера. Метахроматическая лейкодистрофия. Множественная сульфатазная недостаточность.
- E75.4 Липофусциноз нейронов. Болезнь: Баттена, Бильшовского — Янского, Куфса, Шпильмейера — Фогта
- E75.5 Другие нарушения накопления липидов. Болезнь Вольмана.
- E76 Нарушения обмена гликозаминогликанов
- E76.0 Мукополисахаридоз, тип I. Синдромы: «Гурлер», «Гурлер — Шейе», «Шейе».
- E76.1 Мукополисахаридоз, тип II Синдром Хантера
- E76.2 Другие мукополисахаридозы. Недостаточность β-глюкуронидазы. Мукополисахаридозы типов III, IV, VI, VII. Синдром: Марото — Лами (лёгкий), (тяжёлый), Моркио(-подобный), (классический), Санфилиппо (тип A) (тип В) (тип С) (тип D)
- E77 Нарушения обмена гликопротеинов.
- E77.0 Дефекты посттрансляционной модификации лизосомных ферментов. Муколипидоз II (I-клеточная болезнь), Муколипидоз III (псевдополидистрофия Гурлер).
- E77.1 Дефекты деградации гликопротеидов. Аспартилглюкозаминурия. Фукозидоз. Маннозидоз. Сиалидоз (муколипидоз I)
| Лизосомные болезни накопления объединяют группу редких наследственных метаболических заболеваний[2][5]: | |||
|---|---|---|---|
| Болезнь | Хромосома (ген) | Дефицитный фермент | Накапливающиеся субстраты |
| GM1 ганглиозидоз | 3p21.3 (GLB1) | β-Галактозидаза | Ганглиозид GM1, гликопротеины, кератансульфат. |
| Болезнь Тея — Сакса с вариантами, GM2 ганглиозидоз | 15q23-24 (HEXA) | β-Гексозаминидаза A[англ.] | Ганглиозид GM2 |
| Синдром Сандхоффа, GM2 ганглиозидоз | 5q13 (HEXB) | β-Гексозаминидаза A и B | Ганглиозид GM2, глобозид |
| Болезнь Краббе, галактозилцерамидный липидоз | 14q31 (GALC) | Галактозилцерамид-β-галактозидаза | Увеличение отношения галактозоцереброзид/сульфатид |
| Метахромная лейкодистрофия, сульфатидный липидоз | 22q13.3 (ARSA) | Арилсульфатаза A (цереброзидсульфатаза) | Галактозилсульфатиды |
| Недостаточность белкового активатора распада сфинголипида 1, Болезнь Ниманна — Пика, сфингомиелиновый липидоз | 11p15.4-p15.1 (SMPD1) 18q11-q12 (NPC1) 1424.3 (NPC2) | Сфингомиелиназа (?) У некоторых больных специфические изоферменты | Сфингомиелин |
| Болезнь Гоше, глюкозилцерамидный липидоз | 1q22 (GBA) | β-глюкоцереброзидаза | Глюкозилцерамид |
| Болезнь Фабри, тригексозилцерадоз | Xq22 (GLA) | α-Галактозидаза A | Тригексозилцерамид |
| Недостаточность кислой липазы (болезнь Вольмана) | 10q23.2-23.3 (LIPA) | Кислая липаза | Эфиры холестерина, триглицериды |
| Болезнь Фарбера, недостаточность церамидазы | 8p22-p21.3 (ASAH) (ASAH1) | Церамидаза | Церамид |
| Болезнь Помпе, гликогеноз 2 типа | 17q25.2-3 (GAA) | Кислая мальтаза | Гликоген |
| Недостаточность кислой фосфатазы | 11p11.2[31] (ACP2[32]) | Кислая фосфатаза | (?) |
| Фукозидоз | 1p34 (FUCA1) | α-фукозидаза[англ.] | Гликопептиды, гликолипиды, олигосахариды |
| α-маннозидоз | 19cen-q12 (MAN2B1) | α-маннозидаза[англ.] | Олигосахариды |
| β-маннозидоз | 4q22-q25 (MANBA) | β-маннозидаза[англ.] | Олигосахариды |
| Аспартилглюкозаминурия | 4q34.3[33] (AGA)[34] | Аспартилглюкозаминамидаза[англ.] | Аспартилглюкозамин, гликопептиды |
| Мукополисахаридоз IH и IS | 4q16.3 | α-L-идуронидаза[англ.] | Дерматансульфат, гепарансульфат[англ.] |
| Болезнь Хантера, мукополисахаридоз II | Xq27.3-28 | Идуроносульфатсульфатаза | Дерматансульфат, гепарансульфат |
| Синдром Санфилиппо A, мукополисахаридоз IIIA | 17q25.3 | Гепаран-N-сульфатаза (сульфамидаза) | Гепарансульфат |
| Синдром Санфилиппо B, мукополисахаридоз IIIB | 17q21 | N-Ацетил-α-глюкозаминидаза | |
| Синдром Санфилиппо C, мукополисахаридоз IIIC | 8p11.1 | Ацетил-КоА: α-глюкозаминид-N-ацетилтрансфераза | |
| Синдром Санфилиппо D, мукополисахаридоз IIID | 12q14 | N-Ацетилглюкозамин-6-сульфатсульфатаза | |
| Синдром Моркио, мукополисахаридоз IV | 16q24.3 | N-Ацетилгалактозамин-6-сульфатсульфатаза | Кератансульфат |
| Синдром Марото — Лами мукополисахаридоз VI | 5q11-13 (ARSB) | N-Ацетилгексозамин-4-сульфатсульфатаза (арилсульфатаза В) | Дерматансульфат |
| Недостаточность β-глюкуронидазы, мукополисахаридоз VII | 7q21.1-11 | β-глюкуронидаза | Дерматансульфат гепарансульфат (?) |
| Множественная сульфатазная недостаточность | 3p26 (SUMF1) | Арилсульфатазы A, B и C, другие сульфатазы | Сульфатиды, мукополисахариды |
| Сиалидоз (муколипидоз I) | 6p21.3 | Гликопротеиннейраминидаза (сиалидаза) | Сиалилолигосахариды |
| I-клеточная болезнь (муколипидоз II) | 12q23.3 (GNPTAB) | УДФ-N-ацетилглюкозамин-(GlcNAc): гликопротеин-GlcNaCI-фосфотрансфераза | Гликопротеины, гликолипиды |
| Псевдополидистрофия Гурлер (муколипидоз III) | 12q23.3 (GNPTAB) | УДФ-N-ацетилглюкозамин-(GlcNAc): гликопротеин-GlcNaCI-фосфотрансфераза | Гликопротеины, гликолипиды |
| Сиалолипидоз (муколипидоз IV) | 19q13.3-p13.2 (MCOLN1) | Муколипин-1 (Mucolipin-1) | Гликопротеины, гликолипиды |
| Цистиноз | 17p13 (CTNS) | транспортёр цистина англ. Cystin-Transporter | Цистин |
| Восковидные липофусцинозы нейронов | |||
| тип 1 | 1p32 (CLN1) | Пальмитоил-тио-эстераза | «Воск», «липофусцин» |
| тип 2 | 11p15.5 (TPP1) | Трипептидил-пептидаза 1 | |
| тип 3 | 16p12.1 (CLN3) | (?) | |
| тип 4 | (CLN6[35], DNAJC5) | (?) | |
| тип 5 | 13q21.1-q32 (CLN5) | (?) | |
| тип 6 | 15q21-q23 (CLN6) | (?) | |
| тип 7 | 4q28.1-q28.2 (MFSD8) | (?) | |
| тип 8 | 8p23 (CLN8) | (?) | |
| тип 9 | (?) | Дигидро-церамид-синтаза[36] | |
| тип 10 | 11p15.5 (CTSD) | (?) | |
Диагностика
Разработаны специальные методы диагностики, опирающиеся на ряд постоянных признаков, характеризующих болезни накопления липидов:
- накопление в тканях сложных липидов, структурным компонентом которых является церамид;
- скорость синтеза запасаемого липида сравнима со скоростью его биосинтеза у здоровых людей;
- на фоне заболевания наблюдается недостаток специфичного фермента в лизосомах, необходимого для гидролиза липида;
- степень снижения активности фермента во всех тканях одинакова.
Отныне стало возможным выявление в популяции гетерозиготных носителей дефектных генов, ответственных за развитие данных заболеваний, а также выявлять сфинголиподистрофию у плода[4].
Клиническая картина
Предпосылками для проявления лизосомных болезней накопления являются различные генетические дефекты, которые ведут к развитию ферментопатии — недостаточности определённых ферментов, расщепляющих некоторые макромолекулы на уровне внутриклеточных органелл (лизосом). Лизосомные болезни накопления характеризуются[28]:
- прогрессирующим течением,
- высокой инвалидизацией,
- высокой смертностью пациентов.
Наиболее характерными общими особенностями клинической картины для большинства лизосомных болезней накопления являются:
- органомегалия (преимущественно гепатомегалия и спленомегалия),
- костные аномалии,
- различной степени выраженности нарушения со стороны центральной нервной системы,
- грубые особенности строения волос и лица.
Лечение
До недавнего времени терапия наследственных болезней накопления носила исключительно паллиативный характер. Развитие науки позволило с 90-х годов XX столетия приступить к клинической коррекции лизосомных болезней накопления методом эффективной и безопасной ферментозаместительной терапии (англ. Enzyme Replacement Therapy). Принцип данного метода терапии сводится к введению в организм пациента модифицированной формы скомпрометированного генетической патологией фермента, обладающего нормальной активностью. Модификация формы дефектного фермента способствует повышенной проницаемости его в клетки тканей-мишеней, где непосредственно осуществляется процесс катализа реакции гидролиза субстратов накопления. Однако, в связи с коротким (несколько десятков часов) периодом полужизни фермента в клетке, ферментозаместительную терапию необходимо проводить на протяжении всей жизни пациента[1].
См. также
Примечания
- ↑ 1 2 Внутренние болезни: учебник: в 2 т. / под ред. В. С. Моисеева, А. И. Мартынова, Н. А. Мухина. - 3-е изд., испр. и доп. - 2013. - Т.2. - 896 с.: ил. ЧАСТЬ XIII. Наследственные болезни накопления. vmede.org. Дата обращения: 5 ноября 2014. Архивировано 5 ноября 2014 года.
- ↑ 1 2 3 Т. Р. Харрисон. Внутренние болезни в 10 книгах. Книга 8. Пер. с англ. М., [[Медицина (издательство)|Медицина]], 1996, 320 с.: ил. Глава 316. Лизосомные болезни накопления (с. 250—273). med-books.info. Дата обращения: 12 ноября 2014. Архивировано 7 июня 2015 года.
- ↑ 1 2 Биофайл (научно-информационный журнал): Лизосомные болезни накопления. biofile.ru. Дата обращения: 5 ноября 2014. Архивировано 5 ноября 2014 года.
- ↑ 1 2 База знаний по биологии человека: Лизосомные болезни (болезни лизосомного накопления). humbio.ru. Дата обращения: 5 ноября 2014. Архивировано 20 октября 2014 года.
- ↑ 1 2 3 Справочник Т. Р. Харрисона по внутренним болезням, 1992—1997: Глава 316. Лизосомные болезни накопления. rusmedserver.ru. Дата обращения: 5 ноября 2014. Архивировано 3 декабря 2014 года.
- ↑ Лизосомные болезни накопления: Болезнь Гоше. womanadvice.ru. Дата обращения: 5 ноября 2014. Архивировано 5 ноября 2014 года.
- ↑ Evans P. R. Tay-Sachs disease: a centenary (англ.) // Archives of disease in childhood. — 1987. — Vol. 62, no. 10. — P. 1056—1059. Архивировано 8 ноября 2014 года.
- ↑ Gaucher PCE. De l'epithelioma primitif de la rate, hypertrophie idiopathique de la rate sans leucemie [academic thesis] (фр.). — Paris, France, 1882.
- ↑ Pompe J.C. Over idiopathische hypertrophie van het hart // Ned. Tijdschr. Geneeskd. — 1932. — Т. 76. — С. 304—312.
- ↑ Turk V. Special issue: Proteolysis 50 years after the discovery of lysosome in honor of Christian de Duve (англ.) // Biochim Biophys Acta. — 2012. — Vol. 1824, no. 1. — P. 1—2. Архивировано 24 сентября 2015 года.
- ↑ Klionsky D. J. Autophagy revisited: A conversation with Christian de Duve (англ.) // Autophagy[англ.]. — Taylor & Francis, 2012. — Vol. 4, no. 6. — P. 740—743.
- ↑ Berthet, J. Scientific work of Christian de Duve (неопр.) // Bulletin et Memoires de l'Academie Royale de Medecine de Belgique. — 2007. — Т. 162, № 10—12. — С. 499—504. — PMID 18557391. (англ.)
- ↑ Courtoy, P. A tribute to Professor Christian de Duve on his 90th birthday (англ.) // Journal of Cellular and Molecular Medicine[англ.] : journal. — 2007. — Vol. 11, no. 5. — P. 902—905. — doi:10.1111/j.1582-4934.2007.00118.x. — PMID 17979871. (англ.)
- ↑ Tricot, J.P. Nobel prize winner Christian de Duve. From insulin to lysosomes (англ.) // Hormones (Athens, Greece) : journal. — 2006. — Vol. 5, no. 2. — P. 151—155. — doi:10.14310/horm.2002.11179. — PMID 16807228. (англ.)
- ↑ Raju, T.N. The Nobel chronicles. 1974: Albert Claude (1899-1983), George Emil Palade (b 1912), and Christian Réne de Duve (b 1917) (англ.) // The Lancet : journal. — Elsevier, 1999. — Vol. 354, no. 9185. — P. 1219. — doi:10.1016/S0140-6736(05)75433-7. — PMID 10513750. (англ.)
- ↑ Bowers, W.E. Christian de Duve and the discovery of lysosomes and peroxisomes (англ.) // Trends[англ.] : journal. — 1998. — Vol. 8, no. 8. — P. 330—333. — doi:10.1016/S0962-8924(98)01314-2. — PMID 9704410. (англ.)
- ↑ Berthet, J. Introduction of Professor Christian De Duve, Nobel Prize in Medicine and Physiology in 1974 (англ.) // Bulletin et Memoires de l'Academie Royale de Medecine de Belgique : journal. — 1994. — Vol. 149, no. 12. — P. 476—480. — PMID 8563687. (англ.)
- ↑ Takano, T. Profile of Dr. C. De Duve, the 1974 Nobel prize winner in medical physiology (англ.) // Tanpakushitsu Kakusan Koso. Protein, Nucleic Acid, Enzyme : journal. — 1975. — Vol. 20, no. 1. — P. 77—8. — PMID 1094499. (англ.)
- ↑ James, J. The Nobel Prize in Medicine for Claude, Palade and De Duve (англ.) // Nederlands Tijdschrift Voor Geneeskunde : journal. — 1974. — Vol. 118, no. 52. — P. 1949—1951. — PMID 4612387. (англ.)
- ↑ Olsen, BR; Lie, S.O. Nobel prize in medicine 1974 (Albert Claude, George Palade, Christian de Duve) (нид.) // Tidsskrift for den Norske Laegeforening : Tidsskrift for Praktisk Medicin, ny Raekke. — 1974. — Bd. 94, nr. 34—36. — P. 2400—2403. — PMID 4614493. (англ.)
- ↑ Florkin, M. Homage to Albert Claude and Christian de Duve, Nobel Prize laureates in medicine and physiology, 1974 (англ.) // Archives Internationales de Physiologie et de Biochimie : journal. — 1974. — Vol. 82, no. 5. — P. 807—815. — doi:10.3109/13813457409072328. — PMID 4142698. (англ.)
- ↑ De Duve, C; Hooft, C. Quinquennial prizes of the medical sciences, period 1961-1965. Address by Prof. Chr. De Duve (англ.) // Verhandelingen - Koninklijke Vlaamse Academie voor Geneeskunde van Belgie : journal. — 1968. — Vol. 30, no. 7. — P. 381—388. — PMID 5712764. (англ.)
- ↑ Zetterström R. A. A. Claude (1899-1983), C. De Duve (1917-) and G. E. Palade (1912-): Nobel Prize for discoveries in integrated cell physiology. Clarification of aetiology and pathogenesis of a great number of diseases (англ.) // Acta Paediatrica. — 2006. — Vol. 95, no. 12. — P. 1523—1525. — doi:10.1080/08035250601089116.
- ↑ Hers H. G. α-Glucosidase deficiency in generalized glycogen-storage disease (Pompe's disease) (англ.) // Biochemical Journal. — 1963. — Vol. 86, no. 1. — P. 11—16. — PMID 13954110. Архивировано 21 января 2022 года.
- ↑ 1 2 3 la Marca G. Lysosomals // Physician's Guide to the Diagnosis, Treatment, and Follow-Up of Inherited Metabolic Diseases / N. Blau, M. Duran, K. M. Gibson, C. D. Vici. — Springer Berlin Heidelberg, 2014. — P. 785-793. — ISBN 978-3-642-40336-1. (англ.)
- ↑ Winchester B., Vellodi A., Young E. The molecular basis of lysosomal storage diseases and their treatment (англ.) // Biochemical Society Transactions. — 2000. — Vol. 28, no. 1. — P. 150—154. — PMID 10816117.
- ↑ Горбунова, Баранов, 1997, с. 212.
- ↑ 1 2 3 4 Здоров'я України: номер 5/1, март 2009 года, стр. 12-15. Инсульт у молодых пациентов / Редкие наследственные синдромы, сопровождающиеся развитием инсульта. health-ua.com. Дата обращения: 5 ноября 2014. Архивировано 8 декабря 2014 года.
- ↑ Kollmann K. et al. Mannose phosphorylation in health and disease (англ.) // European journal of cell biology. — 2010. — Vol. 89, no. 1. — P. 117—123. Архивировано 24 сентября 2015 года.
- ↑ Parkinson-Lawrence E. J. et al. Lysosomal Storage Disease: Revealing Lysosomal Function and Physiology (англ.) // Physiology. — 2010. — Vol. 25, no. 2. — P. 102—115. Архивировано 7 марта 2016 года.
- ↑ 200950 (англ.)
- ↑ 171650 (англ.)
- ↑ Genetics home referenc: (англ.). AGA. ghr.nlm.nih.gov. Дата обращения: 24 января 2015. Архивировано 28 января 2015 года.
- ↑ OMIM 208400
- ↑ Arsov T. et al. Kufs Disease, the Major Adult Form of Neuronal Ceroid Lipofuscinosis, Caused by Mutations in CLN6 (англ.) // American Journal of Human Genetics. — 2011. — Vol. 88, no. 5. — P. 566—573. Архивировано 24 сентября 2015 года.
- ↑ Schulz A. et al. The CLN9 Protein, a Regulator of Dihydroceramide Synthase (англ.) // J. Biol. Chem. — 2006. — Vol. 281, no. 5. — P. 2784—2794. Архивировано 18 июня 2014 года.
Литература
- Горбунова В. Н., Баранов В. С. . Метаболические дефекты лизосомных ферментов. Болезни накопления. // Введение в молекулярную диагностику и генотерапию наследственных заболеваний. — СПб.: Специальная литература, 1997. — 287 с. — 5000 экз. — ISBN 5-87685-076-4. — С. 206—215.
- Harrison’s Principles of Internal Medicine.