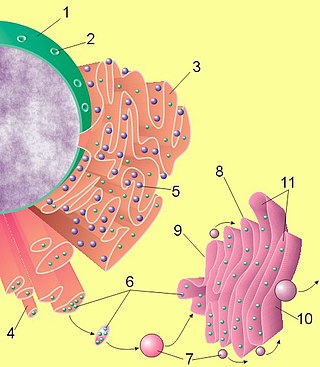
Липогенез
Липогенез — процесс, посредством которого ацетил-КоА превращается в жирные кислоты. Ацетил-КоА представляет собой промежуточную стадию метаболизма простых сахаров, например таких как глюкоза. Посредством липогенеза и последующего синтеза триглицеридов организм эффективно запасает энергию в виде жиров.
Липогенез включает как процесс синтеза жирных кислот, так и синтез триглицеридов (где жирная кислота этерифицированная до глицерина)[1]. Продукты секретируются из печени в виде липопротеинов очень низкой плотности (VLDL). Далее частицы VLDL всасываются непосредственно в кровь, где они созревают и функционируют для доставки эндогенных липидов в периферические ткани.
Синтез жирных кислот
Синтез жирных кислот начинается с ацетил-КоА и накапливается путем добавления двухуглеродных единиц. Синтез происходит в цитоплазме клетки, в отличие от окисления, которое происходит в митохондриях. Многие из ферментов синтеза жирных кислот образуют мультиферментный комплекс под названием синтаза жирной кислоты[2]. Основные производители жирных кислот — это жировая ткань и печень[3].
Регуляция
Гормональная регуляция
Инсулин является пептидным гормоном, который имеет решающее значение при регуляции метаболизма. Инсулин выделяется поджелудочной железой, когда уровень сахара в крови повышается, и это влечёт за собой множество эффектов, которые в целом способствуют поглощению и хранению сахаров, включая липогенез.
Инсулин стимулирует липогенез, главным образом, путем активации двух ферментативных путей. Пируватдегидрогеназа (PDH) превращает пируват в ацетил-КоА. Ацетил-СоА-карбоксилаза (АКК) превращает ацетил-СоА, продуцируемый PDH, в малонил-CoA. Малонил-CoA обеспечивает двухуглеродистые строительные блоки, которые используются для создания более крупных жирных кислот.
Инсулиновая стимуляция липогенеза также происходит путем стимулирования поглощения глюкозы жировой тканью. Увеличение поглощения глюкозы может происходить за счет использования переносчиков глюкозы, направленных на плазматическую мембрану, или путем активации липогенных и гликолитических ферменты, посредством ковалентной модификации[4].
Было обнаружено, что инсулин оказывает долгосрочное влияние на экспрессию липогенных генов. Предполагается, что этот эффект происходит через фактор транскрипции SREBP-1, где ассоциация инсулина и SREBP-1 приводит к экспрессии гена глюкокиназы[5].
Предполагается, что взаимодействие глюкозы и липогенной экспрессии гена управляется увеличением концентрации неизвестного метаболита глюкозы через активность глюкокиназы.
Также на липогенез может влиять другой гормон — лептин (посредством SREBP-1). Он участвует в этом процессе, ограничивая хранение жира за счет ингибирования приема глюкозы и вмешиваясь в другие жировые метаболические пути. Ингибирование липогенеза происходит посредством пониженной регуляции экспрессии гена жирной кислоты и триглицеридов[6].
Было обнаружено, что посредством стимулирования окисления жирных кислот и ингибирования липогенеза лептин контролирует выделение запасённой глюкозы из жировых тканей.
Другие гормоны, которые препятствуют стимуляции липогенеза в жировых клетках, это гормоны роста. Они приводят к потере жира, но стимулируют усиление мышц[7]. Один из предполагаемых механизмов работы гормонов роста заключается в том, что данные гормоны влияют на сигнализацию инсулина, тем самым снижая чувствительность к инсулину и, в свою очередь, регулируют экспрессию синтазы жирных кислот[8].
Другое предположение заключается в том, что гормоны роста имеют механизм фосфорилирования с STAT5A и STAT5B, транскрипционным фактором s, которые являются частью семейства Signal Transducer And Activator Of Transcription (STAT) [9].
Имеются также данные, свидетельствующие о том, что стимулирующий ацилирование белок (ASP) способствует агрегации триглицеридов в жировых клетках[10]. Такая агрегация триглицеридов происходит за счёт увеличения продукции самих триглицеридов[11].
Транскрипционное регулирование
Было обнаружено, что SREBP оказывают гормональные эффекты на экспрессию липогенных генов[12].
SREBP-2 имеет четко определенный режим действия различных членов этого транскрипционного семейства. При высоких уровнях свободного холестерина в клетке, SREBP-2 обнаруживается связанным с эндоплазматическим ретикулумом как незрелый прекурсор. Когда уровень холестерина падает, SREBP-2 протеолитически расщепляется, высвобождая зрелый фрагмент, чтобы он мог перемещаться в ядро и связываться с элементом ответа стерола в промоторной области генов-мишеней. Эти гены затем активируются для транскрипции.
Показано, что SREBP-2 способствует экспрессии генов, участвующих в метаболизме холестерина в клетках печени. Также известно, что SREBP-1 играет роль в активации генов, связанных с липогенезом в печени. Исследования показали, что избыточное экспрессия SREBP-1a или SREBP-1c в клетках печени мышей приводит к накоплению триглицеридов в печени и более высоким уровням экспрессии липогенных генов[13].
Экспрессия липогенных генов в печени через глюкозу и инсулин контролируется SREBP-1[14].
Влияние глюкозы и инсулина на транскрипционный фактор может происходить через различные пути. Имеются данные, свидетельствующие о том, что инсулин способствует экспрессии мРНК SREBP-1 в адипоцитах[15] и гепатоцитах[16].
Также было высказано предположение, что инсулин увеличивает транскрипционную активацию SREBP-1 через MAP-киназ-зависимое фосфорилирование независимо от изменений уровней мРНК[17].
Доказано, что наряду с инсулином глюкозой повышается активность SREBP-1 и экспрессия мРНК[18].
Дефосфорилирование PDH
Инсулин стимулирует активность пируватдегидрогеназы фосфатазы. Фосфатаза удаляет фосфат из пируватдегидрогеназы, активируя его и позволяя превращать пируват в ацетил-СоА. Этот механизм приводит к увеличению скорости катализа этого фермента, поэтому увеличивает уровень ацетил-СоА. В свою очередь, повышенние уровня ацетил-CoA увеличивает не только синтез жира, но также затрагивает и синтез лимонной кислоты.
Ацетил-СоА-карбоксилаза
Инсулин влияет на АКК аналогично PDH. Это приводит к его дефосфорилированию посредством активации PP2A-фосфатазы, активность которого приводит к активации фермента. Глюкагон оказывает антагонистическое действие и увеличивает фосфорилирование, дезактивацию, тем самым ингибируя АКК и замедляя синтез жира.
Воздействие АКК влияет на скорость превращения ацетил-СоА в малонил-СоА. Повышенный уровень малонил-СоА сдвигает баланс в сторону увеличения биосинтеза жирных кислот. Жирные кислоты с длинной цепью являются отрицательными аллостерическими регуляторами АКК, поэтому, когда клетка имеет достаточно жирных кислот с длинной цепью, они в конечном итоге будут ингибировать активность АКК и прекращать синтез жирных кислот.
Концентрации AMP и АТФ клетки работают как индикаторы потребностей АТФ в клетке. Когда АТФ истощается, происходит скачок количества 5’AMP. Это повышение активирует AMP-активированную протеинкиназу, которая фосфорилирует АКК и тем самым ингибирует синтез жира. Это позволяет избежать механизмов запасания глюкозы, в моменты низкого уровня энергии.
АКК также активируется цитратом. Когда в цитоплазме клеток для синтеза жира имеется большое количество ацетил-СоА, она протекает с соответствующей скоростью.
Примечание. Исследования показывают, что метаболизм глюкозы (конкретный метаболит пока точно не определен), помимо влияния инсулина на липогенные ферментные гены, может индуцировать генные продукты для пируваткиназы печени, ацетил-СоА-карбоксилазы и синтазы жирных кислот. Эти гены индуцируются факторами транскрипции ChREBP / Mlx посредством высоких уровней глюкозы в крови[19]. Инсулин-индукция SREBP-1c также участвует в метаболизме холестерина.
Этерификация жирных кислот
Эксперименты проводились для изучения in vivo общей специфики механизмов, участвующих в сложении хиломикронового холестерина и триглицеридов при абсорбции жира у крыс.
Cмеси, содержащие одинаковые количества двух, трех или четырех С14-меченых жирных кислот (пальмитиновая, стеариновая, олеиновая и линолевая кислоты), но с различными соотношениями немеченых жирных кислот, были даны желудочной интубацией для крыс с канюлированными грудными протоками. Полученный таким образом липид хила или хиломикрона хроматографировали на колонках кремниевой кислоты для разделения эфиров холестерина и глицеридов (последний составлял 98,2 % триглицеридов).
После анализа каждого класса липидов для общей радиоактивности использовали газожидкостную хроматографию для измерения общей массы и распределения массы и радиоактивности в отдельных жирных кислотных компонентах каждой липидной фракции. Таким образом была рассчитана удельная радиоактивность каждой жирной кислоты в каждой фракции.
Данные предоставили количественную информацию об относительной специфичности включения каждой жирной кислоты в каждый класс липидов хиломикрона и относительной степени, до которой каждая жирная кислота в каждой липидной фракции была разбавлена эндогенной жирной кислотой. За исключением небольшой дискриминации в отношении стеариновой кислоты, процессы поглощения жирных кислот и образования триглицеридов хиломикрона не проявляют специфичности для одной жирной кислоты по сравнению с другой. Напротив, образование сложного эфира холестерина хиломикрона показало значительную специфичность для олеиновой кислоты по сравнению с остальными тремя жирными кислотами. Эта специфичность не была существенно изменена путем изменения состава тестовой еды, включая холестерин в тестовой муке или путем кормления животного высокохолестериновой диетой в течение нескольких недель, предшествующих исследованию. Наблюдалось значительное разведение диетических жирных кислот с эндогенными жирными кислотами. В одном эксперименте 43 % жирных кислот триглицеридов хиломикрона имели эндогенное происхождение. Относительно больше (54 %) жирных кислот эфира холестерина имеет эндогенное происхождение[20].
Примечания
- ↑ Kersten S. Mechanisms of nutritional and hormonal regulation of lipogenesis (англ.) // EMBO Rep.[англ.] : journal. — 2001. — April (vol. 2, no. 4). — P. 282—286. — doi:10.1093 / embo-reports / kve071. — PMID 11306547. — PMC 1083868.
- ↑ Elmhurst College. Lipogenesis. Дата обращения: 22 декабря 2007. Архивировано 21 декабря 2007 года.
- ↑ J. Pearce. Fatty acid synthesis in liver and adipose tissue (неопр.) // Proceedings of the Nutrition Society. — 1983. — Т. 2. — С. 263—271. — doi:10.1079/PNS19830031.
- ↑ Assimacopoulos-Jeannet, F.; Brichard, S.; Rencurel, F.; Cusin, I.; Jeanrenaud, B. In vivo effects of hyperinsulinemia on lipogenic enzymes and glucose transporter expression in rat liver and adipose tissues (англ.) // Metabolism: Clinical and Experimental[англ.] : journal. — 1995. — 1 February (vol. 44, no. 2). — P. 228—233. — ISSN 0026-0495. — PMID 7869920.
- ↑ Foretz, M.; Guichard, C.; Ferré, P.; Foufelle, F. Sterol regulatory element binding protein-1c is a major mediator of insulin action on the hepatic expression of glucokinase and lipogenesis-related genes (англ.) // Proceedings of the National Academy of Sciences of the United States of America : journal. — 1999. — 26 October (vol. 96, no. 22). — P. 12737—12742. — ISSN 0027-8424. — PMID 10535992. — PMC 23076.
- ↑ Soukas, A.; Cohen, P.; Socci, N. D.; Friedman, J. M. Leptin-specific patterns of gene expression in white adipose tissue (англ.) // Genes & Development : journal. — 2000. — 15 April (vol. 14, no. 8). — P. 963—980. — ISSN 0890-9369. — PMID 10783168. — PMC 316534.
- ↑ Etherton, T. D. The biology of somatotropin in adipose tissue growth and nutrient partitioning (англ.) // The Journal of Nutrition[англ.] : journal. — 2000. — 1 November (vol. 130, no. 11). — P. 2623—2625. — ISSN 0022-3166. — PMID 11053496.
- ↑ Yin, D.; Clarke, S. D.; Peters, J. L.; Etherton, T. D. Somatotropin-dependent decrease in fatty acid synthase mRNA abundance in 3T3-F442A adipocytes is the result of a decrease in both gene transcription and mRNA stability (англ.) // The Biochemical Journal[англ.] : journal. — 1998. — 1 May (vol. 331 ( Pt 3)). — P. 815—820. — ISSN 0264-6021. — PMID 9560309. — PMC 1219422.
- ↑ Teglund, S.; McKay, C.; Schuetz, E.; van Deursen, J. M.; Stravopodis, D.; Wang, D.; Brown, M.; Bodner, S.; Grosveld, G. Stat5a and Stat5b proteins have essential and nonessential, or redundant, roles in cytokine responses (англ.) // Cell : journal. — Cell Press, 1998. — 29 May (vol. 93, no. 5). — P. 841—850. — ISSN 0092-8674. — PMID 9630227.
- ↑ Sniderman, A. D.; Maslowska, M.; Cianflone, K. Of mice and men (and women) and the acylation-stimulating protein pathway (англ.) // Current Opinion in Lipidology : journal. — Lippincott Williams & Wilkins[англ.], 2000. — 1 June (vol. 11, no. 3). — P. 291—296. — ISSN 0957-9672. — PMID 10882345.
- ↑ Murray, I.; Sniderman, A. D.; Cianflone, K. Enhanced triglyceride clearance with intraperitoneal human acylation stimulating protein in C57BL/6 mice (англ.) // American Physiological Society[англ.] : journal. — 1999. — 1 September (vol. 277, no. 3 Pt 1). — P. E474—480. — ISSN 0002-9513. — PMID 10484359.
- ↑ Hua, X; Yokoyama, C; Wu, J; Briggs, M R; Brown, M S; Goldstein, J L; Wang, X. SREBP-2, a second basic-helix-loop-helix-leucine zipper protein that stimulates transcription by binding to a sterol regulatory element. (англ.) // Proceedings of the National Academy of Sciences of the United States of America : journal. — 1993. — 15 December (vol. 90, no. 24). — P. 11603—11607. — ISSN 0027-8424. — PMID 7903453. — PMC 48032.
- ↑ Horton, J. D.; Shimomura, I. Sterol regulatory element-binding proteins: activators of cholesterol and fatty acid biosynthesis (англ.) // Current Opinion in Lipidology : journal. — Lippincott Williams & Wilkins[англ.], 1999. — 1 April (vol. 10, no. 2). — P. 143—150. — ISSN 0957-9672. — PMID 10327282.
- ↑ Shimano, H.; Yahagi, N.; Amemiya-Kudo, M.; Hasty, A. H.; Osuga, J.; Tamura, Y.; Shionoiri, F.; Iizuka, Y.; Ohashi, K. Sterol regulatory element-binding protein-1 as a key transcription factor for nutritional induction of lipogenic enzyme genes (англ.) // The Journal of Biological Chemistry : journal. — 1999. — 10 December (vol. 274, no. 50). — P. 35832—35839. — ISSN 0021-9258. — PMID 10585467.
- ↑ Kim, J B; Sarraf, P; Wright, M; Yao, K M; Mueller, E; Solanes, G; Lowell, B B; Spiegelman, B M. Nutritional and insulin regulation of fatty acid synthetase and leptin gene expression through ADD1/SREBP1. (англ.) // Journal of Clinical Investigation : journal. — 1998. — 1 January (vol. 101, no. 1). — P. 1—9. — ISSN 0021-9738. — doi:10.1172/JCI1411. — PMID 9421459. — PMC 508533.
- ↑ Foretz, Marc; Pacot, Corinne; Dugail, Isabelle; Lemarchand, Patricia; Guichard, Colette; le Lièpvre, Xavier; Berthelier-Lubrano, Cécile; Spiegelman, Bruce; Kim, Jae Bum. ADD1/SREBP-1c Is Required in the Activation of Hepatic Lipogenic Gene Expression by Glucose (англ.) // Molecular and Cellular Biology : journal. — 1999. — 1 May (vol. 19, no. 5). — P. 3760—3768. — ISSN 0270-7306. — PMID 10207099. — PMC 84202.
- ↑ Roth, G.; Kotzka, J.; Kremer, L.; Lehr, S.; Lohaus, C.; Meyer, H. E.; Krone, W.; Müller-Wieland, D. MAP kinases Erk1/2 phosphorylate sterol regulatory element-binding protein (SREBP)-1a at serine 117 in vitro (англ.) // The Journal of Biological Chemistry : journal. — 2000. — 27 October (vol. 275, no. 43). — P. 33302—33307. — ISSN 0021-9258. — doi:10.1074/jbc.M005425200. — PMID 10915800.
- ↑ Hasty, A. H.; Shimano, H.; Yahagi, N.; Amemiya-Kudo, M.; Perrey, S.; Yoshikawa, T.; Osuga, J.; Okazaki, H.; Tamura, Y. Sterol regulatory element-binding protein-1 is regulated by glucose at the transcriptional level (англ.) // The Journal of Biological Chemistry : journal. — 2000. — 6 October (vol. 275, no. 40). — P. 31069—31077. — ISSN 0021-9258. — doi:10.1074/jbc.M003335200. — PMID 10913129.
- ↑ Work from Howard Towle, Catherine Postic, and K. Uyeda.
- ↑ Karmen, Arthur; Whyte, Malcolm; Goodman, DeWitt S. Fatty acid esterification and chylomicron formation during fat absorption: 1. Triglycerides and cholesterol esters (англ.) // The Journal of Lipid Research[англ.] : journal. — 1963. — July (vol. 4). — P. 312—321. — PMID 14168169.