Рили́н — белок, содержащийся в мозге и в других тканях и органах тела человека и других животных. Этот гликопротеин выполняет множество функций, важнейшей из которых является регулировка миграции и позиционирования нервных стволовых клеток в период фетального и раннего послеродового развития, необходимая для нормального формирования коры и других структур головного мозга. Во взрослом мозге рилин регулирует позиционирование нейронов, образуемых в процессе взрослого нейрогенеза, а также вносит вклад в работу механизмов памяти и обучения, модулируя синаптическую пластичность, усиливая и поддерживая долговременную потенциацию, стимулируя развитие дендритов и дендритных шипиков.

Рецептор липопротеинов очень низкой плотности по структуре схож с рецептором липопротеинов низкой плотности, но не способен связывать липопротеины низкой плотности. Рецептор VLDLR играет роль в метаболизме липопротеинов очень низкой плотности. Экспрессия VLDLR высока в сердце, скелетных мышцах, жировой ткани; VLDLR совместно с рецептором LDLR связывает и захватывает остаточные липопротеины: липопротеины промежуточной плотности и хиломикронные ремнанты.
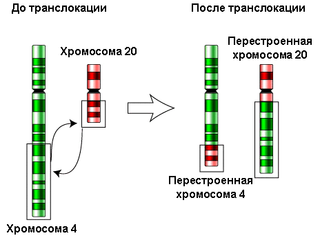
Транслока́ция — тип хромосомных мутаций, при которых происходит перенос участка хромосомы на негомологичную хромосому. Отдельно выделяют реципрокные транслокации, при которых происходит взаимный обмен участками между хромосомами, и Робертсоновские транслокации, или центрические слияния, при которых происходит слияние акроцентрических хромосом с полной или частичной утратой материала коротких плеч.
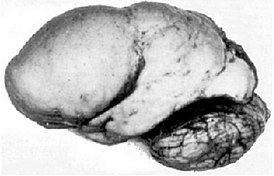
Лиссенцефалия 2, более известная как синдром Норман — Робертс — редкая форма лиссэнцефалии с гипоплазией мозжечка, впервые описанная канадскими врачами Маргарет Норман и Морин Робертс в 1976 году. Причиной заболевания считается мутация гена RELN, кодирующего белок рилин.

Y-хромосо́ма — одна из двух половых хромосом в системе хромосомного определения пола XY, которая встречается у многих животных, в том числе у большинства млекопитающих, включая человека. У млекопитающих содержит ген SRY, определяющий мужской пол организма, а также гены, необходимые для нормального формирования сперматозоидов. Мутации в гене SRY могут привести к формированию женского организма с генотипом XY. Y-хромосома человека состоит из более чем 62 миллионов пар нуклеотидов.

Синдро́м Праде́ра — Ви́лли — редкое наследственное заболевание, причиной которого является отсутствие отцовской копии участка хромосомы 15q11-13. В этом участке хромосомы 15 находятся гены, в регуляции которых задействован геномный импринтинг. Большинство случаев являются спорадическими, для редких описанных семейных случаев характерно неменделевское наследование.

Синдром Ангельмана — обусловленная генетической аномалией патология, характеризующаяся такими признаками, как задержка психического развития, нарушения сна, припадки, хаотические движения, частый смех или улыбки. Также эту болезнь называют «синдром Петрушки» или «синдром счастливой куклы».
Синдро́м лиссэнцефали́и Ми́ллера — Ди́кера — редкое нарушение развития по причине делеции нескольких генов в локусе 17p13. Нарушение развития мозга проявляется в лиссэнцефалии и снижении числа кортикальных слоёв с шести до четырёх. Также заметно изменение формы лица, отмечается замедленный рост, множественные патологии сердца, почек, ЖКТ.
Хромосомные перестройки — тип мутаций, которые изменяют структуру хромосом. Классифицируют следующие виды хромосомных перестроек: делеции, инверсии, дупликации, транслокации, а также дицентрические и кольцевые хромосомы. Известны также изохромосомы, несущие два одинаковых плеча. Если перестройка изменяет структуру одной хромосомы, то такую перестройку называют внутрихромосомной, если же двух разных, то межхромосомной. Хромосомные перестройки подразделяют также на сбалансированные и несбалансированные. Сбалансированные перестройки не приводят к потере или добавлению генетического материала при формировании, поэтому их носители, как правило, фенотипически нормальны. Несбалансированные перестройки меняют дозовое соотношение генов, и, как правило, их носительство сопряжено с существенными отклонениями от нормы.
Синдром Свайера, XY дисгенезия гонад, женская гонадальная дисгенезия или гонадальная дисгенезия — генетическое нарушение, вариант гипогонадизма с кариотипом 46,XY. Организм человека с синдромом Свайера имеет характерный для мужского организма набор хромосом, но половые железы представляют собой гонадный тяж и не производят гормоны. В результате он имеет женские гениталии, женскую репродуктивную систему и выглядит как женщина. В период полового созревания развитие вторичных половых признаков не происходит и наблюдается аменорея. Существует практика удаления гонад в раннем возрасте с целью предотвращения развития рака.

DIDMOAD-синдро́м — аутосомно-рецессивно наследуемый синдром, ассоциированный с инсулинозависимым сахарным диабетом и прогрессирующей атрофией диска зрительного нерва, которые выявляют до 16-летнего возраста. Сочетается с двусторонней прогрессирующей нейросенсорной тугоухостью, несахарным диабетом центрального генеза, дисфункцией автономной нервной системы, приводящей к развитию нейропатического мочевого пузыря и другим проявлениям нейродегенерации, включающими мозжечковую атаксию, миоклональную эпилепсию и атрофию ствола головного мозга. Развёрнутая клиническая картина (фенотипически) встречается приблизительно у 75 % пациентов. Сахарный диабет неаутоиммунного генеза, клинические проявления недостаточности инсулина проявляются приблизительно в 6-летнем возрасте. Средняя продолжительность жизни достигает 30 лет, в течение этого срока происходит развитие полного фенотипа данного синдрома.
Синдро́м де ля Шапе́ля — назван в честь исследователя, впервые охарактеризовавшего его в 1972 году. Синдром относится к редкой хромосомной патологии, возникающей в результате кроссинговера между X- и Y-хромосомами в процессе мейоза, в результате чего одна или обе X-хромосомы содержат нормальный мужской ген SRY. Распространённость данного синдрома составляет 4—5 на 100 000, что меньше встречаемости синдрома Клайнфельтера.
Система антигенов Kell — группа антигенов на поверхности эритроцитов, являющихся важными детерминантами крови и служащих мишенью для многих аутоиммунных или аллоиммунных заболеваний, уничтожающих эритроциты. Показатель Kell обозначается как K, k и Kp. Антигены Kell представляют собой пептиды, обнаруженные в белке kell, 93 кДа трансмембранной цинко-зависимой эндопептидазе, которая отвечает за расщепление эндотелина-3.

X-сцепленное рецессивное наследование — один из видов сцепленного с полом наследования. Такое наследование характерно для признаков, гены которых расположены на Х-хромосоме и которые проявляются только в гомозиготном или гемизиготном состоянии. Такой тип наследования имеет ряд врождённых наследственных заболеваний у человека, эти заболевания связаны с дефектом какого-либо из генов, расположенных на половой Х-хромосоме, и проявляются в случае, если нет другой Х-хромосомы с нормальной копией того же гена. В литературе встречается сокращение XR для обозначения X-сцепленного рецессивного наследования.

Синдром Сандхоффа — наследственное заболевание, вариант 0 GM2-ганглиозидоза из группы лизосомных болезней накопления — нарушение липидного обмена, вызванное наследственным дефицитом (недостаточностью) ферментов бета-гексозаминидазы А и Б.
Синдром Коккейна , также называемый синдром Нил-Дингуолл — редкое аутосомно-рецессивное, нейродегенеративное расстройство, характеризующееся недостатком роста, нарушением развития нервной системы, аномальной чувствительностью к солнечному свету (фотосенсибилизация), заболеваниями глаз и преждевременным старением. Нездоровый вид и неврологические расстройства являются критериями для диагностики, а светочувствительность, нарушения слуха и ненормальные глаза — другие весьма общие черты. Возможны проблемы любого или всех внутренних органов. Это связано с группой расстройств, называемых лейкодистрофия. В основе расстройства лежит дефект механизма репарации ДНК. Интересно, в отличие от других дефектов репарации ДНК, пациенты с CS не предрасположены к раку или инфекции. Синдром Коккейна редок, но это разрушительная болезнь, которая, как правило, приводит к смерти в первом или втором десятилетии жизни. Мутация специфических генов в синдроме Коккейна известна, но широко распространенные эффекты и его отношения с репарацией ДНК еще не очень хорошо исследованы.

УДФ-глюкуронозилтрансфераза 1-1, также известная как UGT-1A, является ферментом, кодируемым геном UGT1A1.

В нейроанатомии изви́лина — выступы (складки), лежащие между бороздами (углублениями) на поверхности полушарий конечного мозга. Борозды и извилины создают характерную «морщинистую» поверхность полушарий головного мозга человека и некоторых других млекопитающих.
DDX3X-синдром — генетическое заболевание, развивающееся преимущественно у лиц женского пола. У пациентов наблюдается задержка умственного развития, признаки аутизма, СДВГ, сниженный мышечный тонус. Причина развития синдрома — мутации, затрагивающие ген DDX3X, расположенный на X-хромосоме. Клиническая картина зависит от характера конкретной мутации.
Аутосомно-доминантная недостаточность ГТФ-циклогидролазы 1 — заболевание, вызываемое нарушением работы фермента ГТФ-циклогидролаза 1, играющего важную роль в цепочке синтеза тетрагидробиоптерина. Это состояние является одной из нескольких известных причин недостаточности тетрагидробиоптерина, а также самой распространённой причиной дофа-зависимой дистонии. У пациентов наблюдается генерализованная дистония с изменением тяжести симптомов в течение дня, а назначение леводопы приводит к радикальному улучшению состояния.