
Биполя́рное расстро́йство, либо биполя́рное аффекти́вное расстро́йство, акр. БАР; либо маниака́льно-депресси́вное расстро́йство; ранее маниака́льно-депресси́вный психо́з, МДП; первоначально циркуля́рный психо́з или циклофрени́я — эндогенное психическое расстройство, проявляющееся в виде аффективных состояний: маниакальных и депрессивных, а нередко и смешанных состояний. Возможны многообразные варианты смешанных состояний.

Га́бсбурги — одна из наиболее могущественных монарших династий Европы на протяжении Средневековья и Нового времени. С 1342 года непрерывно правили в Австрии, с 1438 года занимали престол Священной Римской империи, после роспуска которой (1806) возглавляли многонациональные Австрийскую (1804—1867) и Австро-Венгерскую империи (1867—1918). Правители всех перечисленных государств имели резиденцию в венском Хофбурге.

Фра́нц Ио́сиф I — император Австрии и король Богемии со 2 декабря 1848 года, Апостолический король Венгрии со 2 декабря 1848 по 14 апреля 1849 года и с 13 августа 1849. С 15 марта 1867 года — глава двуединого государства — Австро-Венгерской монархии. С 1 мая 1850 года по 24 августа 1866 года был также президентом Германского союза.

Карл I — император Австро-Венгрии, король Чехии и Венгрии с 21 ноября 1916 года по 12 ноября 1918 года.
Кератоконус — дегенеративное невоспалительное заболевание глаза, при котором роговица истончается и принимает коническую форму. Кератоконус может привести к серьёзному ухудшению зрения. Чаще всего пациенты предъявляют жалобы на светобоязнь, двоение, размазывание изображения. Заболевание является наиболее распространённой формой дистрофии роговицы. Кератоконус поражает примерно одного человека из тысячи, независимо от национальности и места проживания. Диагноз обычно ставится в юности, а наиболее тяжёлой стадии течение болезни достигает к двадцати или тридцати годам.

Карл VII Альбрехт — курфюрст Баварии с 26 февраля 1726 года. Во время войны за австрийское наследство — король Чехии и император Священной Римской империи.

В списке представлены главы государства Австрии со времени, когда страна получила формальную организацию после передачи её территорий Леопольду I императором Священной Римской империи Оттоном II 21 июля 976 года.

Рак моло́чной железы́ — злокачественная опухоль железистой ткани (Аденокарцинома) молочной железы.

Невринома слухового нерва — доброкачественная опухоль-невринома, растущая из шванновских клеток слухового нерва, VIII пары черепных нервов. Ежегодно данная патология выявляется у 1 человека на 100 тысяч населения.
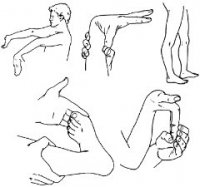
Синдром Э́лерса — Данло́са — это группа наследственных системных дисфункций соединительной ткани, вызванных дефектом в синтезе коллагена. В зависимости от отдельной мутации, серьёзность синдрома может измениться от умеренного до опасного для жизни. Лечения нет, но существует терапия (уход), смягчающая последствия.

Гетерохромия — различный цвет радужной оболочки правого и левого глаза или разная окраска различных участков радужной оболочки одного глаза. Является результатом относительного избытка или недостатка меланина (пигмента). Также под гетерохромией может подразумеваться различная окраска кожи или волосяного покрова.

Краниофарингио́ма — врождённая опухоль головного мозга эпителиального строения, которая развивается из эмбриональных клеток гипофизарного хода. В большинстве случаев доброкачественная, озлокачествление происходит чрезвычайно редко.

Эпендимома — опухоль центральной нервной системы, которая развивается из клеток эпендимы желудочков мозга и центрального канала спинного мозга. Встречается как у детей, так и у взрослых. Чаще всего располагается в задней черепной ямке.

Медуллобластома — злокачественная опухоль, которая развивается из эмбриональных клеток. Первичный узел новообразования располагается в задней черепной ямке в области средней линии мозжечка. Преимущественно медуллобластомы встречаются у детей и составляют у них около 20 % всех первичных опухолей центральной нервной системы.

Нейрофиброматоз — заболевание из группы факоматозов. Существует 7 типов нейрофиброматоза.
Мно́жественная эндокри́нная неоплази́я (МЭН) — этот термин объединяет группу наследственных аутосомно-доминантных синдромов, обусловленных опухолями или гиперплазией нескольких эндокринных желез. Возможны также смешанные типы этих синдромов.

Гамарто́ма лёгкого — доброкачественная опухоль лёгких врождённого происхождения, включающая в себя наряду с лёгочной тканью хрящевые, фиброзные, жировые или сосудистые структуры. Гамартома лёгкого, состоящая из тех же компонентов, что и само лёгкое, отличается их неправильным расположением и степенью дифференцировки. Несмотря на устойчивую эпидемиологическую частоту данной нозологической формы, как и ранее, остаётся сложной диагностика гамартомы, дифференциация её с другими объёмными образованиями лёгкого, в первую очередь с первичными и метастатическими злокачественными процессами. Кроме того, и в наше время зачастую общие представления о гамартоме лёгкого среди врачей размыты и нередко неверны.

Нейрофиброматоз I (первого) типа — самое распространённое наследственное заболевание, предрасполагающее к возникновению опухолей у человека. Описан во второй половине XIX века рядом исследователей, в том числе в 1882 году учеником Рудольфа Вирхова Фридрихом фон Реклингхаузеном. Устаревшие названия — болезнь Реклингхаузена, периферический нейрофиброматоз и др. Является аутосомно-доминантным, встречается с одинаковой частотой у мужчин и у женщин, у 1 из 3500 новорождённых. Другие типы нейрофиброматозов характеризуются наличием как сходных проявлений с I типом, так и отличий.
Нейрофиброматоз II (второго) типа (НФ2) — аутосомно-доминантное наследственное заболевание, которое наследуется или возникает спонтанно, характеризующееся образованием множественных доброкачественных опухолей, преимущественно шванном и менингиом, локализующихся в центральной нервной системе и по ходу периферических нервов. Несмотря на доброкачественную природу новообразований медикаментозного лечения этой болезни не существует, более того, пациенты вынуждены подвергаться многократным хирургическим вмешательствам по удалению опухолей, что рано или поздно приводит к глухоте и слепоте. Развитие заболевания связано с повреждением гена NF2. С мутациями в этом гене связано как образование спорадических шванном и менингиом по всему телу, так и развитие опухолей, на первый взгляд не связанных с нейрофиброматозом, например, злокачественных мезотелиом.

Остеохондропласти́ческая трахеобронхопати́я — редко встречающееся хроническое заболевание дыхательных путей, характеризующееся разрастанием хрящевой и/или костной ткани в подслизистом слое трахеи и крупных бронхов с разной степенью сужения их просвета. Впервые описана при аутопсии Карлом Рокитанским в 1855 году. В мировой медицинской литературе к 1947 году было описано лишь 90 наблюдений остеохондропластической трахеобронхопатии, а к 1993 году — 340 описаний этой формы патологии, из которых лишь 3 случая — в русскоязычной медицинской литературе. В связи с поздним развитием клинических проявлений заболевания и значительной трудностью прижизненной диагностики, во многом обусловленной неосведомлённостью большинства врачей, остеохондропластическая трахеобронхопатия в значительном проценте случаев выявляется лишь посмертно при аутопсии. В качестве случайной находки остеохондропластическая трахеобронхопатия может быть диагностирована при интубации трахеи, компьютерной томографии (КТ) органов грудной клетки и фибробронхоскопии, выполняемых по другим показаниям.
