Синдро́м Туре́тта — генетически обусловленное расстройство центральной нервной системы, которое проявляется в любом возрасте и характеризуется множественными двигательными («моторными») тиками и как минимум одним голосовым, появляющимися много раз в течение дня. В американском Диагностическом и статистическом руководстве по психическим расстройствам пятого издания (DSM-5), наряду с остальными тикозными расстройствами, относится к нейроонтогенетическим моторным расстройствам. Довольно часто встречается коморбидное состояние: синдром Туретта с синдромом дефицита внимания и гиперактивности.

Синдром (болезнь) Марфана аутосомно-доминантное заболевание из группы наследственных патологий соединительной ткани. Синдром вызван мутациями генов, кодирующих синтез гликопротеина фибриллина-1, и является плейотропным. Заболевание характеризуется различной пенетрантностью и экспрессивностью. В классических случаях лица с синдромом Марфана высоки (долихостеномелия), имеют удлинённые конечности, вытянутые пальцы (арахнодактилия) и недоразвитие жировой клетчатки. Помимо характерных изменений в органах опорно-двигательного аппарата, наблюдается патология в органах зрения и сердечно-сосудистой системы, что в классических вариантах составляет триаду Марфана.

Синдро́м Сти́венса — Джо́нсона — острое токсико-аллергическое заболевание, основной характеристикой которого выступают высыпания на коже и слизистых оболочках. Данный синдром — злокачественный тип экссудативной эритемы. Вначале возникает сильная лихорадка и боли в мышцах и суставах. Затем отмирание клеток приводит к возникновению достаточно больших пузырей на слизистой оболочке полости рта, горла, глаз, других участках кожи и слизистых оболочек, появляются дефекты кожи, покрытые плёнками серо-белого цвета, трещины, корки из сгустков запёкшейся крови. Повреждение слизистой оболочки рта мешает есть, закрывание рта вызывает сильную боль, что ведёт к слюнотечению. Возникает воспаление слизистых глаз — конъюнктивит. Роговицы подвергаются фиброзу. Мочеиспускание становится затруднённым и болезненным.

Синдро́м Гийе́на — Барре́ — острая аутоиммунная воспалительная полирадикулоневропатия, проявляющаяся вялыми парезами, нарушениями чувствительности, вегетативными расстройствами.

Синдром мышечной скованности — крайне редкий неврологический синдром неясной этиологии, при котором у пациента нарастает общая скованность мышц.
5-я хромосо́ма челове́ка — одна из 23 пар человеческих хромосом. Хромосома содержит около 181 млн пар оснований, что составляет почти 6 % всего материала ДНК человеческой клетки. Являясь одной из самых больших человеческих хромосом, она тем не менее имеет одну из самых низких плотностей генов. Это частично объясняется наличием большого количества бедных генами участков, в которых наблюдается значительный уровень некодирующих консервативных последовательностей, идентичных имеющимся у немлекопитающих позвоночных, что позволяет предположить их функциональную важность. В настоящее время считается, что на 5-й хромосоме находятся от 900 до 1300 генов.
Синдром Торга-Винчестера — синдром, впервые описанный в 1969 году; у носителей которого отмечается спектр проявлений, в том числе сниженный рост вследствие патологии костей и суставов, помутнения роговицы, грубые черты лица, подкожные узелковые утолщения, огрубение кожи, гипертрихоз. Отклонения вызываются разнообразными мутациями гена MMP2. Их разнообразие привело к тому, что они были описаны как три различных синдрома: синдром Торга, синдром Винчестера, NAO-синдром, и лишь в 2006 году объединены под новым общим именем.

Синдро́м Шегре́на — аутоиммунное системное поражение соединительной ткани, проявляющееся вовлечением в патологический процесс желез внешней секреции, главным образом слюнных и слёзных, и хроническим прогрессирующим течением.

Синдром Пата́у — хромосомное заболевание человека, которое характеризуется наличием в клетках дополнительной хромосомы 13.
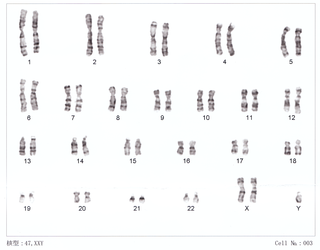
Синдром Клайнфельтера — генетическое отклонение. Клиническая картина синдрома описана в 1942 году в работах Гарри Клайнфельтера и Фуллера Олбрайта. Генетической особенностью этого синдрома является разнообразие цитогенетических вариантов и их сочетаний (мозаицизм). Обнаружено несколько типов полисомии по хромосомам X и Y у лиц мужского пола: 47, XXY; 47, XYY; 48, XXXY; 48, XYYY; 48 XXYY; 49 XXXXY; 49 XXXYY. Наиболее распространён синдром Клайнфельтера. Общая частота его колеблется в пределах 1 на 500—700 новорождённых мальчиков, что делает данный синдром первым по частоте встречаемости среди хромосомных болезней.

Множественная эндокринная неоплазия типа IIb — в данный синдром вошли те же опухоли, что и в МЭН IIa, однако первичный гиперпаратиреоз проявляется реже, а медуллярная карцинома щитовидной железы протекает крайне злокачественно. Выявляется в возрасте 4—37 лет. Отличается от МЭН IIa наличием нейрином (невром) слизистых оболочек и расстройств опорно-двигательного аппарата. Нейриномы чаще локализуются на губах, щеках, языке, других отделах желудочно-кишечного тракта и представляют собой бело-розовые безболезненные узелки диаметром от 1—3 мм до 1 см. Множественная эндокринная неоплазия типа IIb является самым серьёзным вариантом синдрома МЭН.

Синдро́м Ба́рттера — форма гиперальдостеронизма с гиперплазией юкстагломерулярного аппарата почек и резистентностью к сосудосуживающему действию ангиотензина II, обусловленной внешними (вторичными) нарушениями передачи сигнала ангиотензина II.

Синдром коротких рёбер — полидактилии — группа общих генетических патологий человека, характеризующихся короткими рёбрами, укороченными конечностями, полидактилией и смертностью в раннем возрасте вследствие дыхательной недостаточности. Различают четыре подтипа этого синдрома:
- Синдром Салдино — Нунана ;
- Синдром Маевского ;
- Синдром Верма — Наумова ;
- Синдром Бимер — Лангера.
Синдром Макла — Уэльса (MWS) — это мутация в гене CIAS1 с развитием холодового аутовоспалительного синдрома. Является редким аутовоспалительным заболеванием наследственного характера. Преимущественно этническая распространённость — народы Северной Европы. Тип наследования — аутосомно-доминантный. Генетическая основа — мутация гена CIAS1, расположенного на длинном плече 1-й хромосомы (1q44) и кодирующего белка криопирина. Данный белок является основной образуемого в клетке супрамолекулярного комплекса, называемого инфламмасомой, выполняющего функцию превращения pro-IL-1β в активную форму, а также принимающего участие в выполнении программы апоптоза. Холодовой аутовоспалительный синдром тесно связан с двумя другими синдромами: семейной холодовой крапивницы и мультисистемным воспалительным заболеванием неонатального возраста — фактически, все связаны с мутациями гена. В целом Синдром Макла — Уэльса относится к группе криопирин-связанных периодических синдромов (CAPS).
Сухой кератоконъюнктивит, также называемый синдромом сухого глаза или сухим кератитом, представляет собой глазное заболевание, вызываемое сухостью глаз, которая, в свою очередь, вызывается либо пониженной выработкой слёз, либо повышенным испарением слезы. Он обнаружен у человека и некоторых животных. ССГ — это одно из самых распространённых заболеваний, поражающее 5—6 % населения. Частота заболеваемости повышается до 6—9,8 % у женщин в период постменопаузы и составляет целых 34 % у пожилых людей. Фраза «keratoconjunctivitis sicca» — латинская, и её переводом является «сухость (воспаление) роговой оболочки и конъюнктивы».
Окулярный ишемический синдром — созвездие глазных признаков и симптомов вторичных по отношению к тяжёлой, хронической артериальной гипоперфузии глаза. Амавроз слепоты является формой острой потери зрения, вызванной снижением кровотока в глазе, может стать предупреждающим признаком предстоящего инсульта. Следовательно, страдающим временами нечеткостью зрения, следует срочно обратиться к врачу для тщательной оценки сонной артерии. Передний сегмент ишемического синдрома — ишемическая состояние переднего сегмента обычно наблюдался в пост-хирургических случаях. Артериальная окклюзия сетчатки глаза приводит к быстрой гибели клеток сетчатки, в результате чего — серьезной потере зрения.

Мукополисахаридо́з I — группа метаболических заболеваний соединительной ткани, связанных с нарушением обмена кислых гликозаминогликанов, вызванных недостаточностью лизосомного фермента обмена гликозаминогликанов альфа-L-идуронидазы. Данная группа мукополисахаридозов связана с наследственной аномалией, вызванной генетически детерминированным дефектом гена, расположенного в локусе 4q16.3, которая наследуется по аутосомно-рецессивному типу. Проявляется в виде лизосомной болезни накопления, которая приводит к различным дефектам нервной, костной, хрящевой и соединительной тканей.
Синдром Фостер Кеннеди относится к плеяде находок, связанных с опухолями лобных долей.

Слезотечение (Эпифора) — избыточный поток слез на лицо. Клинический признак или порождающее условие — недостаточный дренаж слёзной пленки из глаз, в этом случае слеза будет стекать вниз на лицо, а не через носослёзную систему.

Синдром Дуэйна — врождённый редкий тип косоглазия, чаще всего характеризуется неспособностью глаза двигаться наружу. Синдром был впервые описан офтальмологами Якобом Штиллингом (1887) и Зигмундом Тюрком (1896), а впоследствии стал носить имя Александра Дуэйна, который осветил этот синдром более подробно в 1905 году.