Метахроматическая лейкодистрофия
| Метахроматическая лейкодистрофия | |
|---|---|
 Вид фронтального среза головного мозга при метахроматической лейкодистрофии (МРТ). | |
| МКБ-11 | 5C56.02 |
| МКБ-10 | E75.2 |
| МКБ-10-КМ | E75.25, E75.29 и E75.2 |
| МКБ-9 | 330.0 |
| OMIM | 250100 |
| DiseasesDB | 8080 |
| MedlinePlus | 001205 |
| eMedicine | ped/2893 |
| MeSH | D007966 |
Метахромати́ческая лейкодистрофи́я (англ. Metachromatic leukodystrophy (MLD), также сульфати́дный липидо́з) — редкое наследственное заболевание из группы лизосомных болезней накопления с аутосомно-рецессивным механизмом наследования нарушения обмена веществ[1]. Данная нозологическая единица из разряда лейкодистрофий (патология роста и/или развития миелиновой оболочки, покрывающей большинство нервных волокон центральной и периферической нервной системы) относится к сфинголипидозам. Характеризуется недостаточностью арилсульфатазы А[2] (цереброзидсульфатазы) — фермента лизосом, участвующего в метаболизме сфинголипидов, что вызывает накопление цереброзида сульфата[1][3].
Эпидемиология
Метахроматическая лейкодистрофия встречается с частотой от 1 на 40 000[4] до 1 на 160 000 в зависимости от региона[5].
Наследование

Метахроматическая лейкодистрофия наследуется, как и подавляющее большинство лизосомных болезней накопления, по аутосомно-рецессивному типу наследования[1]. Следовательно, с одинаковой частотой встречается как у мужчин, так и у женщин.
Аутосомно-рецессивный тип наследования на практике означает, что дефектный ген расположен на одной из двух аллельных аутосом. Заболевание клинически манифестирует только в случае, когда обе аутосомы, полученные по одной от отца и матери, являются дефектными по данному гену. Как и во всех случаях аутосомно-рецессивного наследования, если оба родителя несут дефектный ген, то вероятность наследования болезни у потомства составляет 1 из 4. Таким образом в среднем, на одного больного ребёнка в такой семье приходится три без клинических признаков проявлений генной болезни. На схеме синим цветом обозначены здоровые, фиолетовым — носители дефектного гена, красным — метахроматическая лейкодистрофия (два дефектных гена одной аллели). Синим кружочком помечен нормальный ген, красным — дефектный.
Патогенез
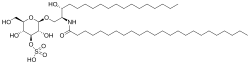

Метахроматическая лейкодистрофия развивается в результате дефицита лизосомного фермента арилсульфатазы А (АРСА, англ. ARSA)[2] или цереброзидсульфатазы. На фоне развёртывания клинической симптоматики уровень активности фермента в лейкоцитах существенно снижается и составляет менее 10 % от нормальных величин[6]. Тем не менее, одного анализа активности цереброзидсульфатазы недостаточно для верификации диагноза. Описана форма псевдо дефицита АРСА (англ. ARSA pseudo deficiency), при которой активность фермента составляет от 5 до 20 % от нормы и при этом отсутствуют признаки метахроматической лейкодистрофии[6]. На фоне дефицита или недостаточности фермента происходит накопление сульфатида во многих тканях организма, что в конечном счёте приводит к разрушению миелиновой оболочки нервных волокон. Миелиновая оболочка — липидное покрытие нервного волокна, которое не только защищает его, но и способствует повышению скорости проведения импульса по нерву. Потеря миелина центральной нервной системой (мозг) и периферической нервной системой нарушает систему контроля и снижает мобильность мышц, таким образом нарушая их функцию.
Арилсульфатаза А (англ. Arylsulfatase A, ARSA) активируется сапозином Б (англ. Saposin B, Sap B) — не ферментативным белковым кофактором. Таким образом, в случае определения накопления сульфатида на фоне нормального уровня арилсульфатазы А, причиной развития метахроматической лейкодистрофии станет не дефицит фермента, а его низкая активность в результате недостаточности сапозина Б. Неактивированный фермент не может достичь нормального уровня эффективности, что в конечном итоге ведёт к накоплению сульфатида, развитию клинической картины и прогрессированию заболевания. Следует отметить, что частота данной патологии значительно ниже, чем традиционной метахроматической лейкодистрофии[7].
Согласно проведенному исследованию утверждается, что накопление сульфатида не вполне отвечает клинической картине метахроматической лейкодистрофии ввиду своей нетоксичности. В связи с этим высказано предположение, что клинические признаки развиваются на фоне накопления лизосульфатида (метаболита сульфатида, лишённого ацильных групп), вещества обладающего цитотоксическими свойствами in vitro[8].
Классификация
Согласно Международной классификации болезней десятого пересмотра (МКБ-10), различают:
- E75 Нарушения обмена сфинголипидов и другие болезни накопления липидов.
Клиническая картина
Двигательные и когнитивные нарушения с судорогами.
Лечение
Лечение не разработано — не существует лекарства от метахроматической лейкодистрофии. Терапия сводится к купированию болевого синдрома и симптомов заболевания. В случае мягких форм болезни, характеризующихся умеренными проявлениями клинической картины, существует возможность трансплантации костного мозга (в том числе, стволовых клеток). Однако, эти методики только разрабатываются и проходят клинические испытания, в результате которых будет выяснена возможность замедления прогрессирования болезни, а также возможность полной остановки развития патологического процесса на уровне клеток центральной нервной системы. Тем не менее, данные полученные в процессе исследования периферической нервной системы не так драматичны, а долгосрочные результаты проведенной терапии на сегодняшний день неоднозначны.
В то же время ведутся разработки других вариантов лечения метахроматической лейкодистрофии. Они включают в себя методы генной терапии, фермент-заместительной терапии (англ. ERT), субстрат-снижающую терапию (с англ. — «СРТ») и повышение активности собственного фермента (англ. EET).
В 2008 году группа международных исследователей при поддержке фонда создала Международный реестр метахроматической лейкодистрофии из научных, академических и отраслевых ресурсов. Задача консорциума создать и управлять общим хранилищем данных, в том числе историй болезни пациентов с сульфатидным липидозом[9].
В 2024 году был одобрен лекарственный препарат для генной терапии лейкодистрофии — атидарсаген аутотемцел[англ.][10].
См. также
Примечания
- ↑ 1 2 3 Le, Tao; Bhushan, Vikas; Hofmann, Jeffrey. First Aid for the USMLE Step 1 (неопр.). — McGraw-Hill Education, 2012. — С. 117. (англ.)
- ↑ 1 2 Poeppel P., Habetha M., Marcão A., Büssow H., Berna L., Gieselmann V. Missense mutations as a cause of metachromatic leukodystrophy, Degradation of arylsulfatase A in the endoplasmic reticulum (англ.) // FEBS Journal[англ.] : journal. — 2005. — March (vol. 272, no. 5). — P. 1179—1188. — doi:10.1111/j.1742-4658.2005.04553.x. — PMID 15720392. (англ.)
- ↑ Metachromatic leukodystrophy at «Dorland’s Medical Dictionary» (англ.)
- ↑ Т. Р. Харрисон. Внутренние болезни в 10 книгах. Книга 8. Пер. с англ. М., [[Медицина (издательство)|Медицина]], 1996, 320 с.: ил. Глава 316. Лизосомные болезни накопления (с. 250—273). med-books.info. Дата обращения: 11 декабря 2014. Архивировано 7 июня 2015 года.
- ↑ Metachromatic leukodystrophy Архивная копия от 30 мая 2019 на Wayback Machine at Genetics Home Reference. Reviewed September 2007 (англ.)
- ↑ 1 2 Fluharty, Arvan. "Arylsulfatase A Deficiency: Metachromatic Leukodystrophy, ARSA Deficiency". GeneReviews, 2006 (англ.)
- ↑ Metachromatic leukodystrophy (MLD) Архивная копия от 22 декабря 2014 на Wayback Machine (англ.)
- ↑ Maria Blomqvist, Volkmar Gieselmann and Jan-Eric Månsson (англ.). Accumulation of lysosulfatide in the brain of arylsulfatase A-deficient mice. lipidworld.com. Дата обращения: 12 декабря 2014. Архивировано 20 декабря 2014 года.
- ↑ MLD Registry http://www.MLDregistry.org Архивная копия от 22 марта 2018 на Wayback Machine (англ.)
- ↑ FDA Approves First Gene Therapy for Children with Metachromatic Leukodystrophy (англ.). FDA (18 марта 2024).
Литература
Ссылки
| Мукополисахаридозы (MPS) |
|
|---|---|
| Муколипидозы (ML) |
|
| Сфинголипидозы | |
| Олигосахаридозы |
|
| Восковидныелипофусцинозынейронов | |
| Прочие | |










