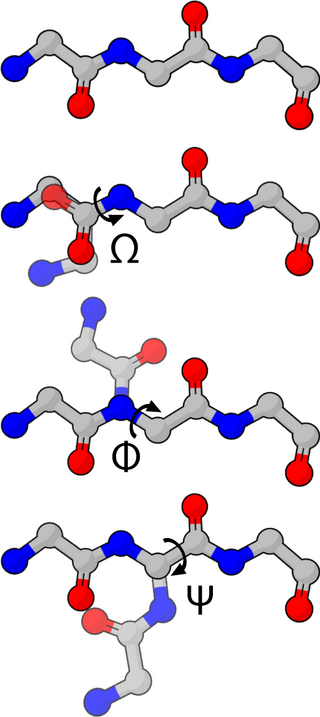
Метод классической молекулярной динамики

Метод молекулярной динамики (метод МД) — метод, в котором временная эволюция системы взаимодействующих атомов или частиц отслеживается интегрированием их уравнений движения[1][2][3]
Основные положения
- Для описания движения атомов или частиц применяется классическая механика. Закон движения частиц находят при помощи аналитической механики.
- Силы межатомного взаимодействия можно представить в форме классических потенциальных сил (как градиент потенциальной энергии системы).
- Точное знание траекторий движения частиц системы на больших промежутках времени не является необходимым для получения результатов макроскопического (термодинамического) характера.
- Наборы конфигураций, получаемые в ходе расчетов методом молекулярной динамики, распределены в соответствии с некоторой статистической функцией распределения, например отвечающей микроканоническому распределению.
Ограничения применимости метода
Метод молекулярной динамики применим, если длина волны Де Бройля атома (или частицы) много меньше, чем межатомное расстояние.
Также классическая молекулярная динамика не применима для моделирования систем, состоящих из легких атомов, таких как гелий или водород. Кроме того, при низких температурах квантовые эффекты становятся определяющими и для рассмотрения таких систем необходимо использовать квантовохимические методы. Необходимо, чтобы времена, на которых рассматривается поведение системы были больше, чем время релаксации исследуемых физических величин.
Временные и пространственные параметры исследуемых систем
Метод классической (полноатомной) молекулярной динамики позволяет с использованием современных ЭВМ рассматривать системы, состоящие из нескольких миллионов атомов на временах порядка нескольких пикосекунд. Применение других подходов (тяжело-атомные, крупно-зернистые (coarse-grained [1]) модели) позволяет увеличить шаг интегрирования и тем самым увеличить доступное для наблюдения время до порядка микросекунд. Для решения таких задач все чаще требуются большие вычислительные мощности, которыми обладают суперкомпьютеры.
История развития метода
Развитие молекулярной динамики шло двумя путями. Первый, обычно называемый классическим, (когда вычисляются траектории атомов) имеет довольно длительную историю. Он восходит к задаче двухчастичного рассеяния, которая может быть решена аналитически. Однако, как хорошо известно, даже уже для трех частиц появляются препятствия, затрудняющие аналитическое решение. Примером может служить простая химическая реакция H + H2 = H2 + H. Для такой реакции Hirschfelder, Eyring, Topley в 1936 году провели попытку расчета нескольких шагов вдоль одной из траекторий. Это было за 30 лет до того, как возможности такого расчета стали возможны на компьютере. Позднее классический подход был подкреплен полуклассическими и квантовохимическими расчетами в тех областях, где влияние квантовых эффектов становилось значимым[4]. Вторым путём развития метода молекулярной динамики стало исследование термодинамических и динамических свойств систем. Идеи, лежащие в основе этого пути восходят к работам Ван-дер-Ваальса и Больцмана.
Следует отметить несколько ключевых работ, определивших развитие метода молекулярной динамики. Первая работа, посвященная моделированию методом молекулярной динамики, вышла в 1957 году. Её авторами были Alder и Waingwright[5]. Целью работы было исследовать фазовую диаграмму системы твердых сфер и в частности области твердого тела и жидкости. В системе твердых сфер частицы взаимодействуют непосредственно при столкновении и двигаются, как свободные частицы между соударениями. Вычисления проводились на компьютерах UNIVAC и на IBM 704.
Статья Dynamics of radiation damage , J.B. Gibson, A. N. Goland, M.Milgram, G.H. Vineyard[6] выполненная в Брукхейвенской национальной лаборатории и появившаяся в 1960 году была, возможно, первым примером моделирования с непрерывным потенциалом. В работе для интегрирования использовался метод конечных разностей. Вычисления проводились на IBM 704 и один шаг занимал около минуты. В статье рассматривалось образование дефектов в меди, вызванных радиационным повреждением. Тема работы была обусловлена проблемами защиты от ядерного нападения.
Aneesur Rahman из Аргоннской национальной лаборатории в своей статье 1964 года Correlation in the motion of atoms in liquid argon[7] изучил свойства жидкого аргона, используя потенциал Леннард-Джонса. Система состояла из 864 атомов. Результаты были получены на компьютере CDC 3600[англ.]. Программный код, использованный для расчетов, лег в основу многих последующих программ.
Loup Verlet вычислил в 1967[8] году фазовую диаграмму аргона, используя потенциал Леннард-Джонса и смоделировал корреляционные функции, чтобы проверить теорию жидкого состояния. В своей работе он разработал процедуру сохранения вычислительных ресурсов, ныне известную как Verlet neighbor list, а также предложил новый метод численного интегрирования уравнений движения.
Применение
Метод молекулярной динамики, изначально разработанный в теоретической физике, получил большое распространение в химии и, начиная с 1970-х годов, в биохимии и биофизике. Он играет важную роль в определении структуры белка и уточнении его свойств (см. также кристаллография, ЯМР). Взаимодействие между объектами может быть описано силовым полем (классическая молекулярная динамика), квантовохимической моделью или смешанной теорией, содержащей элементы двух предыдущих (QM/MM (quantum mechanics/molecular mechanics, QMMM[англ.]).
Наиболее популярными пакетами программного обеспечения для моделирования динамики биологических молекул являются: AMBER, CHARMM (и коммерческая версия CHARMm), GROMACS, GROMOS, LAMMPS, HOOMD-blue и NAMD.
Литература
- Рапапорт Д. К. Искусство молекулярной динамики. — Ижевск: ИКИ, 2012. — 632 с. — ISBN 978-5-4344-0083-1.
- J. A. McCammon, S. C. Harvey (1987) Dynamics of Proteins and Nucleic Acids. Cambridge University Press. ISBN 0-521-35652-0 (paperback); ISBN 0-521-30750-3 (hardback).
- Daan Frenkel, Berend Smit (2001) Understanding Molecular Simulation. Academic Press. ISBN 0-12-267351-4.
- Oren M. Becker, Alexander D. Mackerell Jr, Benoît Roux, Masakatsu Watanabe (2001) Computational Biochemistry and Biophysics. Marcel Dekker. ISBN 0-8247-0455-X.
- Tamar Schlick (2002) Molecular Modeling and Simulation. Springer. ISBN 0-387-95404-X.
Примечания
- ↑ 1. J.M. Haile, Molecular dynamics simulation, Wiley, 1992.
- ↑ M. P. Allen, D. J. D. C. Rapaport The Art of Molecular Dynamics Simulation, 1996.
- ↑ Tildesley Computer simulation of liquids. Oxford University Press, 1989.
- ↑ G.C Schatz, A Kopperman // J. Chem. Phys., v.62 , p.2502, (1975)
- ↑ B.J. Alder, T.E. Waingwright// J. Chem. Phys. v. 27, p.1208, (1957)
- ↑ J.B. Gibson, A. N. Goland, M.Milgram, G.H. Vineyard // Phys Rev, v.120, p.1229, (1960)
- ↑ A Rahman // Phys. Rev. v.136A, p.405, (1964)
- ↑ L. Verlet // Phys Rev, v.159, p.98, (1967)
Ссылки
- The Blue Gene Project Архивная копия от 6 июня 2011 на Wayback Machine (IBM)
- BioSimGrid, a database for biomolecular simulations
- http://www.moldyn.org/library/md/ (недоступная ссылка)