Мотив (молекулярная биология)
Моти́в в молекулярной биологии — относительно короткая последовательность нуклеотидов или аминокислот, слабо меняющаяся в процессе эволюции и, по крайней мере предположительно, имеющая определённую биологическую функцию[1][2][3]. Под мотивом иногда подразумевают не конкретную последовательность, а каким-либо образом описанный спектр последовательностей, каждая из которых способна выполнять определённую биологическую функцию данного мотива[4].
Мотивы встречаются в живых организмах повсеместно и выполняют множество жизненно важных функций, таких как регуляция транскрипции и трансляции (в случае нуклеотидных мотивов), посттрансляционная модификация и клеточная локализация белков, и частично обуславливают их функциональные свойства (лейциновая молния)[2][5]. Они широко используются в биоинформатике для предсказания функций генов и белков, построения карт регуляции, важны для многих задач генной инженерии и молекулярной биологии в целом[6][7][8].
В связи с практической важностью мотивов, разработаны как биоинформатические методы их поиска (MEME, Gibbs Sampler), так и методы поиска мотивов in vivo (ChIP-seq, ChIP-exo). Последние довольно часто дают приблизительные координаты мотивов и их результаты затем уточняются биоинформатическими методами[1][2][6].Для удобства хранения мотивов в базах данных используются их разные, отличающееся степенью детальности, представления, наиболее распространенными из которых являются консенсус и позиционная весовая матрица[2].
Следует отличать мотив от консервативных участков в близкородственных организмах, необладающих значимыми биологическими функциями, где мутационный процесс не успел ещё достаточно их изменить[9].
Мотивы в нуклеиновых кислотах
В случае с ДНК чаще всего мотивы представляют собой короткие последовательности, являющиеся сайтами связывания для белков, таких, как нуклеазы и транскрипционные факторы, или вовлечённые в важные регуляторные процессы уже на уровне РНК, такие как посадка рибосомы, процессинг мРНК и терминация транскрипции[4].
Краткая история изучения
Изучение мотивов в ДНК стало возможным благодаря появлению в 1973 году[10] процедуры секвенирования ДНК (определения последовательности нуклеотидов фрагмента ДНК). Первыми были определены последовательности lac-оператора и лямбда-оператора[11]. Однако до появления более производительных методов секвенирования[12], количество последовательностей мотивов оставалось достаточно малым. К концу 1970-х годов появилось множество примеров мутантных последовательностей (сайтов), связывающих транскрипционные факторы и последовательностей с изменённой специфичностью[13]. С увеличением количества последовательностей, стали развиваться и методы теоретического предсказания мотивов. В 1982 году была впервые сконструирована позиционно-весовая матрица (ПВМ) мотива сайта инициации трансляции. С помощью построенной ПВМ были предсказаны другие сайты инициации трансляции[14]. Этот подход оказался достаточно мощным и до сих пор в разных формах применяется для поиска известных мотивов в геномах, а конкретные методы различаются только видом весовой функции[4]. Однако подход, основанный на построении ПВМ на базе уже имеющихся последовательностей, не позволял находить принципиально новые мотивы, что является более сложной задачей. Первый алгоритм, решавший эту задачу, был предложен Галласом с коллегами в 1985 году[15]. Этот алгоритм был основан на поиске общих слов в наборе последовательностей и давал большой процент ложноотрицательных результатов, однако он стал основой для целого семейства алгоритмов[16]. Позднее были разработаны более точные вероятностные методы: алгоритм MEME, основанный на процедуре максимизации ожидания[17] и алгоритм Gibbs Sampler, также основанный на процедуре максимизации ожидания[18]. Оба метода оказались очень чувствительными и используются в настоящее время для предсказания мотивов в наборах последовательностей.
После разработки мощных средств для предсказания мотивов связывания транскрипционных факторов и установления соответствия между достаточным количеством транскрипционных факторов и мотивов, стало возможным предсказывать функции оперона, лежащего поблизости от мотива по специфичности транскрипционного фактора, с ним связывающегося и наоборот, предсказывать транскрипционный фактор по генам в опероне, лежащем рядом с определённым мотивом[3].
Сайты связывания
Регуляция транскрипции
Характерными примерами регуляции транскрипции, осуществляемой с помощью белка, распознающего специальный мотив, являются:
- Сайт пуринового репрессора PurR у Escherichia coli. PurR связывается с последовательностью в 16 нуклеотидов, которая расположена перед пуриновым опероном и регулирует транскрипцию генов, ответственных за синтез пуриновых и пиримидиновых нуклеотидов[5][19]. Интересно, что у бактерии Bacillus subtilis, эволюционно далёкой от кишечной палочки, также есть пуриновый репрессор, не гомологичный PurR[20];
- Сайт лактозного оперона Lac. Лактозный оперон контролируется репрессором LacI, который, связывая ДНК, препятствует транскрипции генов, ответственных за катаболизм лактозы[6].
Регуляция трансляции
Одними из наиболее известных примеров регуляции трансляции при помощи мотив-распознающих регуляторов являются:
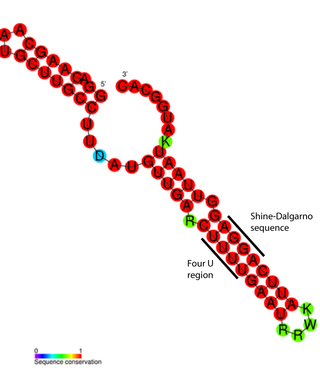
- Сайт посадки рибосомы прокариот — последовательность Шайн — Дальгарно[21], здесь связывание происходит с рибопротеином;
- Сайт посадки рибосомы эукариот — последовательность Козак, связывание происходит с эукариотическим фактором инициации трансляции eIF1[7];
- IRE — регуляторные элементы, располагающиеся на 5’UTR и/или 3’UTR мРНК ферментов (к примеру, ферритина), регулирующие содержание железа в клетке. С этими мотивами связываются белки IRP1 (цитозольная форма аконитазы) и IRP2 (каталитически неактивный гомолог аконитазы), регулируя самим фактом своего связывания с мРНК скорость её деградации или скорость трансляции, происходящей с неё[22].
Сила мотива
Сила взаимодействия белка или РНК с ДНК мотивом зависит в первую очередь от последовательности данного мотива. Различают «сильные» мотивы, дающие сильное взаимодействие с белком или РНК и «слабые» мотивы, с которыми взаимодействие слабее. Практически всегда удаётся получить так называемую «консенсусную последовательность» («консенсус»), то есть такую последовательность, в каждой позиции которой стоит буква, наиболее часто встречающаяся в соответствующей позиции в последовательностях мотивов из разных организмов. Консенсусная последовательность принимается за самую сильную, каковой она почти всегда и является[23]. Более слабые мотивы получаются из неё с помощью небольшого (чаще всего 1—3) числа замен[24].
Эволюция силы мотива
В процессе эволюции сила мотивов регулируется с помощью естественного отбора, причём мотив может становиться как сильнее, так и слабее[25]. Характерным примером такой подстройки силы мотива может служить изменчивость последовательности Шайна — Дальгарно (ШД). Есть тесная корреляция между необходимым организму количеством транслируемого белка и силой ШД перед ним[8].
В случае с ШД, хотя сила связывания белка и напрямую коррелирует с силой связывания 16S-субъединицы рибосомы, в связи с особенностями инициации трансляции, консенсусная последовательность не обязательно будет гарантировать наиболее эффективную трансляцию (из-за затруднённого ухода рибосомы с сайта инициации)[6]. Поэтому последовательность Шайна — Дальгарно чаще всего содержит 4—5 нуклеотидов из консенсусной последовательности при длине последней примерно в 7 нуклеотидов[26].
РНК-переключатели
Не всегда наличие мотива, явно выполняющего биологически значимую роль, влечёт за собой наличие белка-регулятора. Регуляция также может осуществляться за счёт связывания РНК с каким-либо низкомолекулярным веществом. На этом принципе построены РНК-переключатели — структуры, образующиеся на РНК во время транскрипции, способные связывать малые молекулы[27][28]. Связывание молекулы влияет на способность рибопереключателя останавливать транскрипцию или препятствовать трансляции. В этом случае важной оказывается не последовательность нуклеотидов как таковая, а наличие комплементарных нуклеотидов на нужных местах в последовательности[4].
Регуляция за счёт вторичной структуры
Регуляция трансляции также может осуществляться только за счёт образуемой нуклеиновой кислотой вторичной структуры.
- Ро-независимый терминатор транскрипции — шпилька, образующаяся на синтезируемой мРНК до начала трансляции, препятствующая дальнейшему синтезу мРНК (Терминатор (ДНК))[29];
- IRES — сложная структура в мРНК вирусов эукариот, обеспечивающий внутреннюю инициацию трансляции[30].
Структура мотива
Зачастую, мотивы, связывающие транскрипционные факторы, имеют вид прямых повторов некоторой последовательности, обратных повторов или палиндромных последовательностей. Это можно объяснить работой транскрипционных факторов в виде димеров белков, в которых каждый из мономеров связывает одну и ту же последовательность. Встречаются также мотивы большей повторности[6]. Такое строение мотивов обеспечивает большую резкость реакции на изменение внешних условий. К примеру, если связывание зависит от концентрации одного вещества в клетке, то получаем зависимость силы реакции клетки, описываемую уравнением Михаэлиса — Ментен. С увеличением числа связывающихся единиц белка (будем считать, что действие связывания белка с мотивом проявляется только в случае связывания со всеми повторами) зависимость всё больше становится похожей на сигмоиду, в пределе стремясь к функции Хевисайда, описывающей один из главных принципов реагирования живых систем на многие воздействия — закон «всё или ничего» (англ. all-or-nothing law)[6], к примеру, формирования потенциала действия[31].
Мотивы в белках
Для белков следует различать
- мотив в последовательности аминокислот
- структурный мотив — взаимное расположение нескольких близко расположенных элементов вторичной структуры в пространстве[2][22]. На последовательности же эти элементы могут далеко отстоять друг от друга[32].
Мотивы в первичной структуре (последовательности белка)
Мотивы в первичной структуре похожи на мотивы в нуклеиновых кислотах. Характерными примерами таковых являются:
- сигнальные пептиды — короткие аминокислотные последовательности в составе белка длиной порядка 3—60 аминокислот[33], определяющие, в какой компартмент клетки будет отправлен после синтеза. Пример — сигнал ядерной локализации;
- сайты посттрансляционной модификации белков, представляющие собой консервативные пептиды порядка 5—12 аминокислот[6]. Пример — сайты ацетилирования в белке[34]
Структурные мотивы
В белках структурные мотивы описывают связи между элементами вторичной структуры. Такие мотивы часто имеют участки переменной длины, которые в некоторых случаях могут и вовсе отсутствовать[22].
- Лейциновая молния — характерен для димерных белков, связывающих ДНК. Лейциновая молния обеспечивает контакт двух мономеров белка за счёт гидрофобных взаимодействий[22][35]. Для него характерно наличие в каждой седьмой позиции остатка лейцина.
- Цинковые пальцы — характерен для ДНК-связывающих факторов транскрипции[22][36];
- Спираль-поворот-спираль — ДНК-связывающий мотив, именно такой ДНК-связывающий фрагмент у Lac-репрессора[22].
- Гомеодомен — мотив, связывающий ДНК и РНК. У эукариот белки с гомеодоменами индуцируют дифференцировку клеток, запуская каскады генов, необходимых для образования тканей и органов. Похож на мотив «спираль-поворот-спираль», потому часто отдельно не выделяется[22][37].
- Укладка Россмана — мотив, связывающий нуклеотиды (к примеру — НАД)[38]. Встречается, в частности, в дегидрогеназах, в том числе в глицеральдегид-3-фосфатдегидрогеназе, участвующей в гликолизе.
- EF-рука — мотив, связывающий ионы Са2+, также подобен мотиву «спираль-поворот-спираль»[39].
- Гнездо — три последовательных аминокислотных остатка формируют сайт связывания аниона[40].
- Ниша — три последовательных аминокислотных остатка формируют сайт связывания катиона[41].
- Бета-шпилька — два β-тяжа, соединённых коротким разворотом цепи белка[42].
Кроме бета-шпильки выделяют и множество других мотивов, функция которых состоит в формировании структурного каркаса белка[43].
Близким к термину структурный мотив белка является укладка — характерное расположение элементов вторичной структуры. В силу своей схожести термины часто используются один вместо другого и грань между ними размыта[43][44].
Представление мотивов
Изначально имеется набор мотивов из разных последовательностей и ставится задача[2]:
- представить их компактно и наглядно;
- уметь по представлению мотива осуществлять поиск его новых вхождений.
Существует несколько общепризнанных способов представления мотивов[45]. Часть из них подходит как для белков, так и для нуклеотидов, другая часть — только для белков или нуклеотидов.
Консенсус
Строгий консенсус
Строгим консенсусом мотива назовем строчку, состоящую из самых представленных букв в множестве реализаций мотива. На практике, указывается не просто наиболее частая буква в данной позиции, но и, если максимальная частота встречаемости какой-либо буквы в данной позиции меньше заданного порога, то на этом месте в консенсусе ставится x (любая буква алфавита). По такому консенсусу мы почти наверняка находим последовательности, реально являющиеся мотивами, но упускаем большое число мотивов, отличающихся от консенсуса на несколько замен[2][4][9]. Ниже приведён пример строгого консенсуса для участка мотива пяти взятых из UniProt белков с мотивом лейциновой молнии (порог был взят равным 80 %):
| Номер позиции | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| UniProt ID | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 |
| O35048 | L | S | P | C | G | L | R | L | I | G | A | H | P | I | L |
| Q6XXX9 | L | G | Q | D | I | C | D | L | F | I | A | L | D | V | L |
| Q9N298 | L | G | Q | V | T | C | D | L | F | I | A | L | D | V | L |
| Q61247 | L | S | P | L | S | V | A | L | A | L | S | H | L | A | L |
| B0BC06 | L | T | I | G | Q | Y | S | L | Y | A | I | D | G | T | L |
| Консенсус | L | x | x | x | x | x | x | L | x | x | x | x | x | x | L |
Нестрогий консенсус
Нестрогим консенсусом назовем последовательность списков букв, наиболее представленных на соответствующем месте. Описываются все или наиболее часто встречающиеся буквы в данной позиции (обычно устанавливается минимальный порог частоты)[2]. Фактически, мотив описывается при помощи регулярного выражения[4][9]. В качестве обозначений используют:
- Алфавит — совокупность отдельных символов, обозначающих определённую аминокислоту/нуклеотид или набор аминокислот/нуклеотидов;
ABC— строка из символов алфавита, обозначающая последовательность символов, следующих друг за другом;[ABC]— любая строка символов, взятых из алфавита в квадратных скобках соответствует любому из соответствующих символов; например [ABC] соответствует или A или B или C;{ABC..DE}— любая строка символов, взятых из алфавита, соответствует любой аминокислоте, кроме тех, что находятся в фигурных скобках; например{ABC}соответствует любой аминокислоте, кромеA,BиC;xв нижнем регистре — любой символ алфавита.
В случае с таким представлением приходится балансировать между чувствительностью консенсуса (количеством реальных мотивов, которые им получится отыскать) и специфичностью (способностью метода отбраковывать мусорные последовательности)[1]. Ниже приведен пример нестрого консенсуса для тех же пяти последовательностей белков, что и для строго консенсуса (порог был взят равным 20 %). Видим, что в позиции 10 мотив не совсем объективен — лейцин (L) и изолейцин (I) — очень близкие по свойствам аминокислоты, и логично было бы их обе занести в консенсус.
| Номер позиции | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| UniProt ID | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 |
| O35048 | L | S | P | C | G | L | R | L | I | G | A | H | P | I | L |
| Q6XXX9 | L | G | Q | D | I | C | D | L | F | I | A | L | D | V | L |
| Q9N298 | L | G | Q | V | T | C | D | L | F | I | A | L | D | V | L |
| Q61247 | L | S | P | L | S | V | A | L | A | L | S | H | L | A | L |
| B0BC06 | L | T | I | G | Q | Y | S | L | Y | A | I | D | G | T | L |
| Консенсус | L | [SG] | [PQ] | x | x | C | D | L | F | I | A | [LH] | D | V | L |
Prosite-консенсус (для белков)
PROSITE использует ИЮПАК для обозначения однобуквенных кодов аминокислот, за исключением символа конкатенации «-», используемого между элементами паттерна. При использовании PROSITE добавляется несколько символов, облегчающих представление белкового мотива[46]:
- '
<' — шаблон ограничивается N-концом последовательности; - '
>' — шаблон ограничивается C-концом последовательности;
Если e — шаблон элемента, и m и n два десятичных целых числа и m <= n, то:
e(m)эквивалентно повторениюeровноmраз;e(m,n)эквивалентно повторениюeровноkраз для любого целогоkудовлетворяющего условию:m<=k<=n;
Пример: мотив домена с сигнатурой C2H2-type цинкового пальца выглядит следующим образом: C-x(2,4)-C-x(3)-[LIVMFYWC]-x(8)-H-x(3,5)-H[47]
Позиционная весовая матрица
Позиционной весовой матрицей называется такая матрица, столбцы которой соответствуют позиции в последовательности, а строчки соответствуют буквам в алфавите. Значениями этой матрицы являются частоты (или монотонные функции от частот) встречаемости данной буквы в данной позиции на последовательности. При этом обычно, чтобы исключить нулевые частоты к числу встреч каждой буквы позиции добавляют некоторое число, исходя из априорного распределения букв в подобных последовательностях[4] (к примеру, вводят поправку Лапласа[48]). Данный подход, как и предыдущие, неявно предполагает, что позиции в мотиве независимы, чего на самом деле не наблюдается даже для нуклеотидных последовательностей[2][4].
Пусть у нас есть 7 последовательностей ДНК, представляющих собой мотив[9]:
| Номер позиции | ||||||||
|---|---|---|---|---|---|---|---|---|
| Номер последовательности | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
| 1 | A | T | C | C | A | G | C | T |
| 2 | G | G | G | C | A | A | C | T |
| 3 | A | T | G | G | A | T | C | T |
| 4 | A | A | G | C | A | A | C | C |
| 5 | T | T | G | G | A | A | C | T |
| 6 | A | T | G | C | C | A | T | T |
| 7 | A | T | G | G | C | A | C | T |
Позиционная матрица для них будет иметь следующий вид (+1 — учёт правила Лапласа)[9]:
| Номер позиции | ||||||||
|---|---|---|---|---|---|---|---|---|
| Нуклеотид | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
| A | 5 + 1 | 1 + 1 | 0 + 1 | 0 + 1 | 5 + 1 | 5 + 1 | 0 + 1 | 0 + 1 |
| C | 1 + 1 | 0 + 1 | 1 + 1 | 4 + 1 | 2 + 1 | 0 + 1 | 6 + 1 | 1 + 1 |
| G | 0 + 1 | 1 + 1 | 6 + 1 | 3 + 1 | 0 + 1 | 1 + 1 | 0 + 1 | 0 + 1 |
| T | 1 + 1 | 5 + 1 | 0 + 1 | 0 + 1 | 0 + 1 | 1 + 1 | 1 + 1 | 6 + 1 |
Частоты можно пронормировать на общее число последовательность, тем самым получив оценку вероятности встречи данного нуклеотида в данной последовательности (собственно, обычно в таком представлении и хранится PWM)[2]:
| Номер позиции | ||||||||
|---|---|---|---|---|---|---|---|---|
| Нуклеотид | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
| A | 0,55 | 0,18 | 0,09 | 0,09 | 0,55 | 0,55 | 0,09 | 0,09 |
| C | 0,18 | 0,09 | 0,18 | 0,45 | 0,27 | 0,09 | 0,64 | 0,18 |
| G | 0,09 | 0,18 | 0,64 | 0,36 | 0,09 | 0,18 | 0,09 | 0,09 |
| T | 0,18 | 0,55 | 0,09 | 0,09 | 0,09 | 0,18 | 0,18 | 0,64 |
HMM (скрытые марковские модели)

Для большей точности можно учитывать зависимость соседних позиций в мотиве с помощью скрытых марковских моделей первого и более высоких порядков[2][4]. Этот подход сопряжён с некоторыми трудностями, так как для его применения необходимо наличие достаточно представительной выборки вариантов мотивов. В случае предыдущего примера имеем:
- Для марковской модели порядка 0 (вероятность появления нуклеотида в данной позиции от других позиций не зависит — другой способ трактовки PWM)[4];

- Для марковской модели порядка 1 (вероятность появления нуклеотида в данной позиции зависит только от нуклеотида в предыдущей последовательности. Число параметров модели сильно возросло)[4]. При расчёте вероятностей перехода также использовалось правило Лапласа. Эмисионные вероятности для состояний равны 1 для нуклеотидов, которым они соответствуют, 0 — для остальных.
В случае мотивов, содержащих участки переменного размера и нуклеотидного состава, можно было бы вводить отдельно модель для этих участков, отдельно — для консервативных, а затем «склеивать» их в одну модель путём добавления промежуточных «молчащих» состояний и вероятностей перехода в них и из них[4].
СКС (стохастическая контекстно-свободная грамматика)
В случае мотивов, формирующих вторичные структуры (РНК-переключатели) в РНК, в элементах вторичной структуры важно учитывать возможность спаривания нуклеотидов. С этой задачей справляются СКС. Однако обучение СКС требует ещё большего размера выборки, чем HMM, и сопряжено с рядом трудностей[4].
Представление для больших базах данных
В тех случаях, когда важна скорость поиска и допустим пропуск некоторых вхождений нашего мотива, исследователи прибегают к различным уловкам, позволяющим с приемлемой точностью зашифровать пространственную структур биополимера (РНК или белка) путём расширения алфавита[49].
Представление мотивов в белках с помощью кодирования пространственной структуры белка
Оперон Escherichia coli репрессор лактозы LacI (PDB 1lcc chain A) и ген активатор катаболизма (PDB 3gap chain A) оба имеют мотив спираль-поворот-спираль, но их аминокислотные последовательности не очень схожи. Группой исследователей был разработан код, который они назвали «трёхмерный код цепи», представляющий структуру белка в виде строки из писем. Эта схема кодирования, по мнению авторов, показывает сходство между белками гораздо более отчётливо, чем аминокислотные последовательности[49]:
Пример: сравнение двух упомянутых выше белков при помощи этой схемы кодирования[49]:
| PDB ID | 3D-code | Amino acid sequence |
|---|---|---|
1lccA | TWWWWWWWKCLKWWWWWWG | LYDVAEYAGVSYQTVSRVV |
3gapA | KWWWWWWGKCFKWWWWWWW | RQEIGQIVGCSRETVGRIL |
| Сравнение | Видно явное сходство между белками | По аминокислотной последовательности белки сильно отличаются |
где W соответствует α-спирали, и E и D соответствует β-нити.
Представление мотивов в РНК с помощью вторичной структуры (foldedBlast)
В данной работе с целью применения алгоритма поиска, схожего с BLAST, нуклеотидный алфавит (ATGC, так как поиск осуществлялся в геноме) был расширен за счёт комбинирования нуклеотидов и трех символов, характеризующих их предположительное направление спаривания[50]:
(— нуклеотид спарен с нуклеотидом справа;)— нуклеотид спарен с нуклеотидом слева;.— нуклеотид не спарен.
Таким образом получалось 12 букв нового алфавита (4 нуклеотида * 3 «направления»), при правильном использовании позволяющий осуществлять BLAST-подобный поиск, названный авторами foldedBlast[50].
Логотип последовательностей
Для визуального представления мотивов часто используют логотип последовательностей — графического представления консервативности каждой позиции в мотиве. При этом данную визуализацию можно успешно применять как и в случае представления мотива в виде консенсуса или позиционной весовой матрицы, так и для представления HMM модели последовательности, как это сделано в базе белковых семейств Pfam[51].
Кроме того, если использовать, к примеру, яркость каждой нуклеотида в мотиве как индикатор того, насколько часто ему соответствует в этом же мотиве комплементарный нуклеотид, то можно частично представлять и информацию о вторичной структуре мотива. Так сделано, например, в биоинформатическом веб-сервисе RegPredict[52].
Поиск сайтов связывания транскрипционных факторов in silico
В случае поиска в нуклеотидных последовательностях мотивов, отвечающих за связывание регуляторных белков пользуются соображением, что они [мотивы] меняются сравнительно медленно, а значит, если взять организмы, достаточно далёкие друг от друга, чтобы в высоковариабельных позициях их последовательностей успели накопиться мутации, а сайты измениться сильно ещё не успели, то можно пользоваться правилом «что консервативно — то важно»[2]. После получения последовательностей, в которых предполагается наличия специфичного мотива, в основном используют два подхода к поиску последовательности мотива — филогенетический футпринтинг и сведение задачи к задаче поиска вставленного мотива.
Филогенетический футпринтинг
Филогенетический футпринтинг — полуавтоматический метод. Последовательности обрабатываются программой множественного выравнивания, и в получившемся выравнивании исследователем ищутся паттерны, которые можно считать мотивами. Одним из наиболее успешных примеров применения данного подхода можно считать расшифровку способа кодирования нерибосомных пептидов нерибосомными пептид-синтетазами (NRPS)[2][53][54]. Данный метод не позволяет полностью автоматизировать процесс поиска мотивов, но при этом и не имеет столь сильных ограничений, как следующие.
Задача поиска вставленного мотива
В случае с мотивами без (почти без) разрывов и без (почти без) участков переменной длины возможно свести задачу поиска мотива к задаче поиска вставленного мотива (англ. Planted motif search)[2][9].
Формулировка задачи следующая: «На вход предоставлены n строк s1, s2, …, sn длины m, каждая составленная из символов алфавита A, и два числа — l и d. Найдите все строки x длины l такие, что любая из предоставленных строки содержит хотя бы одну подпоследовательность, находящуюся от x на расстоянии Хэмминга не больше d»[55].
Так как в общем случае неизвестно, все ли полученные нами последовательности имеют искомый мотив, а также неизвестна его точная длина, то обычно задачу решают эвристическими методами — максимизируя вероятность найденного мотива при данных последовательностях. На этом принципе построены программы MEME[17] и GibbsSampler[56].
Если задать минимальный порог на число последовательностей, в которых должен содержаться мотив, и как-либо ограничить его длину, то можно использовать и точные способы решения данной задачи, к примеру — алгоритм RISOTTO[57]. Некоторые из них позволяют снимать часть ограничений на искомый мотив — в RISOTTO искомый мотив может иметь разрывы, состоять из нескольких частей.
Однако эти методы редко дают результаты лучше, чем MEME и GibbsSamler, а работают они значительно дольше[2][58].
Поиск сайтов связывания in vitro
ChIP-seq
Метод анализа ДНК-белковых взаимодействий, комбинирующий идеи иммунопреципитации хроматина (ChIP) и высокоэффективном секвенировании ДНК (белок пришивается к ДНК, затем кусочки ДНК, пришившиеся к белку отправляются на секвенирование). В ходе работы метода получаются участки длиной около 150 нуклеотидов, которые затем можно анализировать in silico на наличие мотива[59].
ChIP-on-chip
Как и в случае использования метода ChIP-seq проводится иммунопреципитации хроматина (ChIP), затем сшивка с белком обращается и полученная ДНК гибридизуется с ДНК-микрочипом. Метод ChIP-on-chip дешевле, чем ChIP-seq, однако сильно уступает последнему в точности[6].
ChIP-exo
Также метод, основанный на иммунопреципитации хроматина (ChIP). Использование экзонуклеазы фага λ, деградирующей ДНК только с 5'-конца и только в случае отсутствия контакта с белком, позволяет добиваться точности порядка нескольких нуклеотидов в определении положения сайта связывания белка[60].
SELEX
Итеративный метод поиска нуклеотидных последовательностей, хорошо связывающихся с данным белком[61]. Процедура в общем случае выглядит так:
- Интересующий нас белок пришивается к колонке, через которую далее пропускается раствор с набором последовательностей, состоящих из рандомизированного участка и адаптера;
- Последовательности, задержавшиеся на колонке клонируют процедуре ПЦР, причем состав реакционной смеси подобран таким образом, чтобы вносить дополнительные ошибки при копировании. Полученные клоны отправляются на новый раунд SELEX;
- Через каждые несколько участков условия (pH раствора, его ионная сила) ужесточаются, чтобы на колонке оставались все более и более специфичные к белку последовательности;
- Получающиеся на выходе последовательности часто похожи на реальные мотивы связывания белка в живых организмах.
DamID
Делается гибридный белок из изучаемого белка и адениновой ДНК-метилтрансферазы Dam[62]. В естественных условиях аденин в большинстве эукариот не метилируется. Когда же гибридный белок связывается с каким-либо сайтом в ДНК организма, метилтрансферазная часть модифицирует аденины в районе этого сайта, что позволяет затем с помощью эндонуклеаз рестрикции выделить участок, на котором с большой долей вероятности находится искомый мотив.
Примечания
- ↑ 1 2 3 D'haeseleer Patrik. What are DNA sequence motifs? (англ.) // Nature Biotechnology. — 2006. — 1 April (vol. 24, iss. 4). — P. 423–425. — ISSN 1087-0156. — doi:10.1038/nbt0406-423. Архивировано 12 апреля 2017 года.
- ↑ 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 Compeau Phillip, Pevzner Pavel. Bioinformatics Algorithms: An Active Learning Approach, 2nd Ed. Vol. 1 by Phillip Compeau (англ.). — 2nd edition. — Active Learning Publishers, 2015. — 384 p. — ISBN 9780990374619.
- ↑ 1 2 Koonin Eugene V. The Logic of Chance: The Nature and Origin of Biological Evolution. — 1 edition. — FT Press, 2011-06-23. — 529 с. — ISBN 978-0132542494.
- ↑ 1 2 3 4 5 6 7 8 9 10 11 12 13 Durbin Richard, Eddy Sean R., Krogh Anders, Mitchison Graeme. Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids. — Cambridge University Press, 1998. — 372 с. — ISBN 978-0521620413.
- ↑ 1 2 Purine repressor - Proteopedia, life in 3D (англ.). proteopedia.org. Дата обращения: 11 апреля 2017. Архивировано 12 апреля 2017 года.
- ↑ 1 2 3 4 5 6 7 8 Alberts Bruce, Johnson Alexander, Lewis Julian, Raff Martin, Roberts Keith. Molecular Biology of the Cell. — 4th. — Garland Science, 2002-01-01. — ISBN 0815332181. — ISBN 0815340729. Архивировано 27 сентября 2017 года.
- ↑ 1 2 Pestova T. V., Kolupaeva V. G., Lomakin I. B., Pilipenko E. V., Shatsky I. N. Molecular mechanisms of translation initiation in eukaryotes (англ.) // Proceedings of the National Academy of Sciences of the United States of America. — 2001. — 19 June (vol. 98, iss. 13). — P. 7029–7036. — ISSN 0027-8424. — doi:10.1073/pnas.111145798. Архивировано 23 апреля 2017 года.
- ↑ 1 2 Evfratov Sergey A., Osterman Ilya A., Komarova Ekaterina S., Pogorelskaya Alexandra M., Rubtsova Maria P. Application of sorting and next generation sequencing to study 5΄-UTR influence on translation efficiency in Escherichia coli (англ.) // Nucleic Acids Research. — 2017. — 7 April (vol. 45, iss. 6). — P. 3487–3502. — ISSN 0305-1048. — doi:10.1093/nar/gkw1141. Архивировано 12 апреля 2017 года.
- ↑ 1 2 3 4 5 6 Jones Neil C., Pevzner Pavel A. An Introduction to Bioinformatics Algorithms. — 1 edition. — The MIT Press, 2004. — 435 с. — ISBN 9780262101066.
- ↑ Gilbert W, Maxam A. The nucleotide sequence of the lac operator (англ.) // Proceedings of the National Academy of Sciences. — 1973. — December (vol. 70, iss. 12). — P. 3581—3584. — PMID 4587255. Архивировано 24 апреля 2017 года.
- ↑ Maniatis T, Ptashne M, Backman K, Kield D, Flashman S, Jeffrey A, Maurer R. Recognition sequences of repressor and polymerase in the operators of bacteriophage lambda (англ.) // Cell. — 1975. — June (vol. 5, iss. 2). — P. 109—113. — PMID 1095210. Архивировано 24 апреля 2017 года.
- ↑ Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors (англ.) // Proceedings of the National Academy of Sciences. — 1977. — December (vol. 74, iss. 12). — P. 5463—5467. Архивировано 2 апреля 2017 года.
- ↑ Stormo GD. DNA binding sites: representation and discovery. (англ.) // Bioinformatics. — 2000. — January (vol. 16, iss. 1). — P. 16—23. Архивировано 19 апреля 2017 года.
- ↑ Stormo GD, Schneider TD, Gold LM. Characterization of translational initiation sites in E. coli (англ.) // Nucleic Acids Research. — 1982. — 11 May (vol. 10, iss. 9). — P. 2971—2996. Архивировано 24 апреля 2017 года.
- ↑ Galas DJ, Eggert M, Waterman MS. Rigorous pattern-recognition methods for DNA sequences. Analysis of promoter sequences from Escherichia coli. (англ.) // Journal of Molecular Biology. — 1985. — 5 November (vol. 186, no. 1). — P. 117–128. Архивировано 24 апреля 2017 года.
- ↑ Stormo GD. DNA binding sites: representation and discovery. (англ.) // Bioinformatics. — 2000. — January (vol. 16, no. 1). — P. 16–23. Архивировано 19 апреля 2017 года.
- ↑ 1 2 T. L. Bailey, C. Elkan. The value of prior knowledge in discovering motifs with MEME (англ.) // Proceedings. International Conference on Intelligent Systems for Molecular Biology. — 1995. — 1 January (vol. 3). — P. 21–29. — ISSN 1553-0833. Архивировано 24 апреля 2017 года.
- ↑ Lawrence CE1, Altschul SF, Boguski MS, Liu JS, Neuwald AF, Wootton JC. Detecting subtle sequence signals: a Gibbs sampling strategy for multiple alignment. (англ.) // Science. — 1993. — 8 October (vol. 262, no. 5131). — P. 208–214. Архивировано 24 апреля 2017 года.
- ↑ Jendresen Christian Bille, Martinussen Jan, Kilstrup Mogens. The PurR regulon in Lactococcus lactis - transcriptional regulation of the purine nucleotide metabolism and translational machinery (англ.) // Microbiology (Reading, England). — 2012. — 1 August (vol. 158, iss. 8). — P. 2026–2038. — ISSN 1465-2080. — doi:10.1099/mic.0.059576-0. Архивировано 19 апреля 2017 года.
- ↑ Sinha Sangita C., Krahn Joseph, Shin Byung Sik, Tomchick Diana R., Zalkin Howard. The purine repressor of Bacillus subtilis: a novel combination of domains adapted for transcription regulation (англ.) // Journal of Bacteriology. — 2003. — 1 July (vol. 185, iss. 14). — P. 4087–4098. — ISSN 0021-9193. — doi:10.1128/JB.185.14.4087-4098.2003. Архивировано 19 апреля 2017 года.
- ↑ Shine J., Dalgarno L. Terminal-sequence analysis of bacterial ribosomal RNA. Correlation between the 3'-terminal-polypyrimidine sequence of 16-S RNA and translational specificity of the ribosome (англ.) // European Journal of Biochemistry. — 1975. — 1 September (vol. 57, iss. 1). — P. 221–230. — ISSN 0014-2956. Архивировано 19 апреля 2017 года.
- ↑ 1 2 3 4 5 6 7 Nelson David L., Cox Michael M. Lehninger Principles of Biochemistry. — 7 edition. — W. H. Freeman, 2017-01-01. — 1328 с. — ISBN 9781464126116.
- ↑ Stormo G. D., Schneider T. D., Gold L. Quantitative analysis of the relationship between nucleotide sequence and functional activity (англ.) // Nucleic Acids Research. — 1986. — 26 August (vol. 14, iss. 16). — P. 6661–6679. — ISSN 0305-1048. Архивировано 19 апреля 2017 года.
- ↑ Stormo G. D. DNA binding sites: representation and discovery (англ.) // Bioinformatics (Oxford, England). — 2000. — 1 January (vol. 16, iss. 1). — P. 16–23. — ISSN 1367-4803. Архивировано 19 апреля 2017 года.
- ↑ Shultzaberger Ryan K., Zehua Chen, Lewis Karen A., Schneider Thomas D. Anatomy of Escherichia coli σ 70 promoters (англ.) // Nucleic Acids Research. — 2007. — 1 February (vol. 35, iss. 3). — P. 771–788. — ISSN 1362-4962. — doi:10.1093/nar/gkl956. Архивировано 19 апреля 2017 года.
- ↑ J. Shine, L. Dalgarno. Terminal-sequence analysis of bacterial ribosomal RNA. Correlation between the 3'-terminal-polypyrimidine sequence of 16-S RNA and translational specificity of the ribosome (англ.) // European Journal of Biochemistry. — 1975. — 1 September (vol. 57, iss. 1). — P. 221—230. — ISSN 0014-2956. Архивировано 19 апреля 2017 года.
- ↑ Рибопереключатель, РНК-переключатель (riboswitch). humbio.ru. Дата обращения: 11 апреля 2017. Архивировано 12 апреля 2017 года.
- ↑ Samuel E. Bocobza, Asaph Aharoni. Small molecules that interact with RNA: riboswitch-based gene control and its involvement in metabolic regulation in plants and algae (англ.) // The Plant Journal: For Cell and Molecular Biology. — 2014. — 1 August (vol. 79, iss. 4). — P. 693–703. — ISSN 1365-313X. — doi:10.1111/tpj.12540. Архивировано 19 апреля 2017 года.
- ↑ Hironori Otaka, Hirokazu Ishikawa, Teppei Morita, Hiroji Aiba. PolyU tail of rho-independent terminator of bacterial small RNAs is essential for Hfq action (англ.) // Proceedings of the National Academy of Sciences of the United States of America. — 2011. — 9 August (vol. 108, iss. 32). — P. 13059–13064. — ISSN 0027-8424. — doi:10.1073/pnas.1107050108. Архивировано 3 июля 2022 года.
- ↑ Hiroshi Yamamoto, Marianne Collier, Justus Loerke, Jochen Ismer, Andrea Schmidt. Molecular architecture of the ribosome‐bound Hepatitis C Virus internal ribosomal entry site RNA (англ.) // The EMBO Journal. — 2015. — 14 December (vol. 34, iss. 24). — P. 3042–3058. — ISSN 0261-4189. — doi:10.15252/embj.201592469.
- ↑ Камкин Андрей, Каменский Андрей Александрович. Фундаментальная и клиническая физиология. — Academia, 2004-01-01. — 1072 с. — ISBN 5769516755.
- ↑ "Structural Motifs". EMBL-EBI Train online (англ.). 2011-11-25. Архивировано 12 апреля 2017. Дата обращения: 12 апреля 2017.
- ↑ Gonter Blobel, Bernhand Dobberstein. Transfer of proteins across membranes. I. Presence of proteolytically processed and unprocessed nascent immunoglobulin light chains on membrane-bound ribosomes of murine myeloma (англ.) // The Journal of Cell Biology. — 1975. — 1 December (vol. 67, iss. 3). — P. 835–851. — ISSN 0021-9525. Архивировано 2 апреля 2022 года.
- ↑ Qiu Wang-Ren, Sun Bi-Qian, Xiao Xuan, Xu Zhao-Chun, Chou Kuo-Chen. iPTM-mLys: identifying multiple lysine PTM sites and their different types (англ.) // Bioinformatics (Oxford, England). — 2016. — 15 October (vol. 32, iss. 20). — P. 3116–3123. — ISSN 1367-4811. — doi:10.1093/bioinformatics/btw380. Архивировано 19 апреля 2017 года.
- ↑ Landschulz W. H., Johnson P. F., McKnight S. L. The leucine zipper: a hypothetical structure common to a new class of DNA binding proteins (англ.) // Science (New York, N.Y.). — 1988. — 24 June (vol. 240, iss. 4860). — P. 1759–1764. — ISSN 0036-8075. Архивировано 19 апреля 2017 года.
- ↑ Klug A., Rhodes D. Zinc fingers: a novel protein fold for nucleic acid recognition (англ.) // Cold Spring Harbor Symposia on Quantitative Biology. — 1987. — 1 January (vol. 52). — P. 473–482. — ISSN 0091-7451. Архивировано 19 апреля 2017 года.
- ↑ Bürglin Thomas R., Affolter Markus. Homeodomain proteins: an update (англ.) // Chromosoma. — 2016. — 1 January (vol. 125). — P. 497–521. — ISSN 0009-5915. — doi:10.1007/s00412-015-0543-8. Архивировано 8 марта 2021 года.
- ↑ Rao S. T., Rossmann M. G. Comparison of super-secondary structures in proteins (англ.) // Journal of Molecular Biology. — 1973. — 15 May (vol. 76, iss. 2). — P. 241–256. — ISSN 0022-2836. Архивировано 23 апреля 2017 года.
- ↑ Nelson Melanie R., Thulin Eva, Fagan Patricia A., Forsén Sture, Chazin Walter J. The EF-hand domain: A globally cooperative structural unit (англ.) // Protein Science : A Publication of the Protein Society. — 2017. — 14 April (vol. 11, iss. 2). — P. 198–205. — ISSN 0961-8368. — doi:10.1110/ps.33302.
- ↑ Watson James D., Milner-White E. James. A novel main-chain anion-binding site in proteins: the nest. A particular combination of φ,ψ values in successive residues gives rise to anion-binding sites that occur commonly and are found often at functionally important regions1 (англ.) // Journal of Molecular Biology. — 2002. — 11 January (vol. 315, iss. 2). — P. 171–182. — doi:10.1006/jmbi.2001.5227.
- ↑ Torrance Gilleain M., David P. Leader, Gilbert David R., Milner-White E. James. A novel main chain motif in proteins bridged by cationic groups: the niche (англ.) // Journal of Molecular Biology. — 2009. — 30 January (vol. 385, iss. 4). — P. 1076–1086. — ISSN 1089-8638. — doi:10.1016/j.jmb.2008.11.007. Архивировано 23 апреля 2017 года.
- ↑ Milner-White E. J., Poet R. Four classes of beta-hairpins in proteins. (англ.) // Biochemical Journal. — 1986. — 15 November (vol. 240, iss. 1). — P. 289–292. — ISSN 0264-6021.
- ↑ 1 2 Efimov Alexander V. Favoured structural motifs in globular proteins (англ.) // Structure. — 1994. — 1 November (vol. 2, iss. 11). — P. 999–1002. — doi:10.1016/S0969-2126(94)00102-2.
- ↑ Holm L., Sander C. Dictionary of recurrent domains in protein structures (англ.) // Proteins. — 1998. — 1 October (vol. 33, iss. 1). — P. 88–96. — ISSN 0887-3585. Архивировано 23 апреля 2017 года.
- ↑ Schneider T. D., Stephens R. M. Sequence logos: a new way to display consensus sequences (англ.) // Nucleic Acids Research. — 1990. — 25 October (vol. 18, iss. 20). — P. 6097–6100. — ISSN 0305-1048. Архивировано 20 апреля 2017 года.
- ↑ de Castro Edouard, Sigrist Christian J. A., Gattiker Alexandre, Bulliard Virgini, Langendijk-Genevaux Petra S. ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins (англ.) // Nucleic Acids Research. — 2006. — 1 July (vol. 34, iss. Web Server issue). — P. W362–365. — ISSN 1362-4962. — doi:10.1093/nar/gkl124. Архивировано 6 октября 2016 года.
- ↑ InterPro EMBL-EBI. Zinc finger C2H2-type (IPR013087) < InterPro < EMBL-EBI (англ.). www.ebi.ac.uk. Дата обращения: 15 апреля 2017. Архивировано 15 апреля 2017 года.
- ↑ Флах Петер. Машинное обучение. Наука и искусство построения алгоритмов, которые извлекают знания из данных. Учебник. — ДМК Пресс, 2015-01-01. — 400 с. — ISBN 9785970602737, 9781107096394.
- ↑ 1 2 3 Matsuda H., Taniguchi F., Hashimoto A. An approach to detection of protein structural motifs using an encoding scheme of backbone conformations (англ.) // Pacific Symposium on Biocomputing. Pacific Symposium on Biocomputing. — 1997. — 1 January. — P. 280–291. — ISSN 2335-6936. Архивировано 23 апреля 2017 года.
- ↑ 1 2 Tseng Huei-Hun, Weinberg Zasha, Gore Jeremy, Breaker Ronald r., Ruzzo Walter l. Finding non-coding rnas through genome-scale clustering (англ.) // Journal of bioinformatics and computational biology. — 2017. — 12 April (vol. 7, iss. 2). — P. 373–388. — ISSN 0219-7200.
- ↑ Schuster-Böckler Benjamin, Jörg Schultz, Rahmann Sven. HMM Logos for visualization of protein families (англ.) // BMC Bioinformatics. — 2004. — 1 January (vol. 5). — P. 7. — ISSN 1471-2105. — doi:10.1186/1471-2105-5-7.
- ↑ Novichkov Pavel S., Rodionov Dmitry A., Stavrovskaya Elena D., Novichkova Elena S., Kazakov Alexey E. RegPredict: an integrated system for regulon inference in prokaryotes by comparative genomics approach (англ.) // Nucleic Acids Research. — 2010. — 1 July (vol. 38, iss. Web Server issue). — P. W299–307. — ISSN 1362-4962. — doi:10.1093/nar/gkq531. Архивировано 24 апреля 2017 года.
- ↑ Marahiel Mohamed A. Multidomain enzymes involved in peptide synthesis (англ.) // FEBS Letters. — 1992. — 27 July (vol. 307, iss. 1). — P. 40–43. — ISSN 1873-3468. — doi:10.1016/0014-5793(92)80898-Q. Архивировано 12 апреля 2017 года.
- ↑ Stachelhaus T., Mootz H. D., Marahiel M. A. The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases (англ.) // Chemistry & Biology. — 1999. — 1 August (vol. 6, iss. 8). — P. 493–505. — ISSN 1074-5521. — doi:10.1016/S1074-5521(99)80082-9. Архивировано 19 апреля 2017 года.
- ↑ Keich U., Pevzner P. A. Finding motifs in the twilight zone (англ.) // Bioinformatics (Oxford, England). — 2002. — 1 October (vol. 18, iss. 10). — P. 1374–1381. — ISSN 1367-4803. Архивировано 19 апреля 2017 года.
- ↑ Thompson William A., Newberg Lee A., Conlan Sean, McCue Lee Ann, Lawrence Charles E. The Gibbs Centroid Sampler (англ.) // Nucleic Acids Research. — 2007. — 1 July (vol. 35, iss. Web Server issue). — P. W232–237. — ISSN 1362-4962. — doi:10.1093/nar/gkm265.
- ↑ Carvalho A. M., Freitas A. T., Oliveira A. L., Sagot M. F. An efficient algorithm for the identification of structured motifs in DNA promoter sequences (англ.) // IEEE/ACM Transactions on Computational Biology and Bioinformatics. — 2006. — 1 April (vol. 3, iss. 2). — P. 126–140. — ISSN 1545-5963. — doi:10.1109/TCBB.2006.16. Архивировано 8 сентября 2017 года.
- ↑ Dinh Hieu, Rajasekaran Sanguthevar, Davila Jaime. qPMS7: A Fast Algorithm for Finding (ℓ, d)-Motifs in DNA and Protein Sequences (англ.) // PLOS ONE. — 2012. — 24 July (vol. 7, iss. 7). — ISSN 1932-6203. — doi:10.1371/journal.pone.0041425. Архивировано 15 июня 2022 года.
- ↑ Johnson David S., Mortazavi Ali, Myers Richard M., Wold Barbara. Genome-wide mapping of in vivo protein-DNA interactions (англ.) // Science (New York, N.Y.). — 2007. — 8 June (vol. 316, iss. 5830). — P. 1497–1502. — ISSN 1095-9203. — doi:10.1126/science.1141319. Архивировано 24 апреля 2017 года.
- ↑ Rhee Ho Sung, Pugh B. Franklin. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution (англ.) // Cell. — 2011. — 9 December (vol. 147, iss. 6). — P. 1408–1419. — ISSN 1097-4172. — doi:10.1016/j.cell.2011.11.013. Архивировано 24 апреля 2017 года.
- ↑ Tuerk C., Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase (англ.) // Science (New York, N.Y.). — 1990. — 3 August (vol. 249, iss. 4968). — P. 505–510. — ISSN 0036-8075. Архивировано 24 апреля 2017 года.
- ↑ Greil Frauke, Moorman Celine, van Steensel Bas. DamID: mapping of in vivo protein-genome interactions using tethered DNA adenine methyltransferase (англ.) // Methods in Enzymology. — 2006. — 1 January (vol. 410). — P. 342–359. — ISSN 0076-6879. — doi:10.1016/S0076-6879(06)10016-6. Архивировано 24 апреля 2017 года.
Литература
- Дурбин Р., Эдди Ш., Крог А., Митчисон Г. Анализ биологических последовательностей = Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids. — Регулярная и хаотическая динамика, Институт компьютерных исследований, 2006. — С. 480. — ISBN 5939725597.
- Jones Neil C., Pevzner Pavel A. An Introduction to Bioinformatics Algorithms (англ.). — The MIT Press, 2004. — ISBN 9780262101066.
- Compeau Phillip, Pevzner Pavel. Bioinformatics Algorithms: An Active Learning Approach, 2nd Ed. Vol. 1 by Phillip Compeau (англ.). — Active Learning Publishers, 2015. — P. 384. — ISBN 9780990374619.
- Durbin Richard, Eddy Sean R., Krogh Anders, Mitchison Graeme. Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids (англ.). — Cambridge University Press, 1998. — P. 372. — ISBN 978-0521620413.
- Nelson David L., Cox Michael M. Lehninger Principles of Biochemistry (англ.). — W. H. Freeman, 2017. — P. 1328. — ISBN 9781464126116.
Ссылки
Видеокурсы по данной теме
- Находим скрытые в ДНК сообщения — часть курса по биоинформатике от всемирно известного учёного П. А. Певзнера
Сервисы поиска мотивов
- MEME Suite of motif-based sequence analysis tools — сервис для поиска мотивов в последовательностях одноимённым алгоритмом MEME
- The Gibbs Motif Sampler — сервис для поиска мотивов в последовательностях алгоритмом Gibbs Sampler
- RISOTTO motif discovery tool — главная страница программы для точного поиска мотивов RISOTTO
- PMS — точный поиск мотивов при помощи алгоритмов семейства PMS
- Bioprospector — поиск мотивов в последовательностях алгоритмом Gibbs Sampler
- XXmotif — сервис для поиск мотивов в нуклеотидных последовательностях на основании прямой оптимизации статистической значимости PWM
Базы данных мотивов
- PROSITE — база данных белковых семейств и доменов
- TRANSFAC — коммерческая (ограниченный публичный доступ) база данных транскрипционных факторов
- HOCOMOCO Архивная копия от 6 июня 2013 на Wayback Machine — коллекция траскрипционных факторов человека и мыши
- Minimotif Miner — поиск коротких известных мотивов
Прочее
- Wikiomic Sequence motifs page — статья о мотивах в последовательностях
- Cis-analysis — список и короткие описания части программ поиска мотивов в последовательностях