
Са́харный диабе́т 1-го ти́па — аутоиммунное заболевание эндокринной системы, основным диагностическим признаком которого является хроническая гипергликемия — повышенный уровень сахара в крови, полиурия, как следствие этого — жажда; потеря веса; чрезмерный либо сниженный аппетит; Сильное общее утомление организма; Боли в животе; при длительном проявлении болезни и отсутствии диагностики заболевания, начинается отравление организма продуктами распада жиров - часто проявляется в виде запаха ацетона от кожи, изо рта;

Неса́харный диабе́т — редкое заболевание, связанное с нарушением функции гипоталамуса либо гипофиза, которое характеризуется полиурией и полидипсией (жажда).

Митохондриа́льные заболева́ния — группа наследственных заболеваний, связанных с дефектами в функционировании митохондрий, приводящими к нарушениям энергетических функций в клетках эукариот, в частности, человека. На 2021 год известно более 350 генов, приводящих к таким заболеваниям.
Сахарный диабет взрослого типа у молодых, более известный как MODY-диабет - термин, описывающий несколько схожих по протеканию форм диабета с аутосомно-доминантным типом наследования. Исторически термином MODY обозначали разновидность диабета, при которой заболевание обнаруживается в молодом возрасте, а протекает мягко, подобно "взрослому" диабету второго типа, но зачастую без снижения чувствительности к инсулину. С углублением знаний определение MODY-диабета сузилось, и в новой этиологически-обоснованной классификации MODY относят к типам диабета "связанным с генетическим дефектом функционирования бета-клеток", с разбивкой на подтипы в соответствии с конкретным поражённым геном (MODY1-MODY9).
Митохондриальная тРНК лейцина 1 — тРНК человека, кодируемая митохондриальным геном MT-TL1. Представляет собой молекулу транспортной РНК длиной в 75 нуклеотидов, основной функцией MT-TL1 является перенос остатка аминокислоты лейцина к растущей полипептидной цепочке при трансляции митохондриальных мРНК на рибосомах митохондрий.

Ге́тероплазми́я — различия в последовательности ДНК каких-либо органоидов в одном и том же организме, зачастую даже в одной клетке. Источники гетероплазмии — соматические мутации, гетероплазмия в ооците, либо передача части митохондриальной ДНК по отцовской линии.

Синдром MELAS — прогрессирующее нейродегенеративное заболевание, характеризующееся проявлениями, перечисленными в названии, и сопровождается полиморфной симптоматикой — диабетом, судорогами, снижением слуха, сердечными заболеваниями, низким ростом, эндокринопатиями, непереносимостью физических нагрузок и нейропсихиатрическими отклонениями. В каждом конкретном случае набор симптомов и их тяжесть может сильно отличаться, поскольку синдром связан с мутациями во многих генах: MTTL1, MTTQ, MTTH, MTTK, MTTS1, MTND1, MTND5, MTND6, MTTS2. Мутации могут возникать впервые у конкретного пациента, либо наследоваться по материнской линии. Всего к 2009 году было обнаружено 23 миссенсных точечных мутаций и 4 делеции мтДНК, приводящих к MELAS, однако продолжаются сообщения о новых пациентах с симптомами расстройства при отсутствии известных мутаций.

Синдром MERRF — редкое митохондриальное заболевание, вызываемое мутациями в следующих генах: MTTK, MTTL1, MTTH, MTTS1, MTTS2, MTTF. Симптомы MERRF также проявляются при мутациях гена MTND5.
Не́оната́льный са́харный диабе́т — редко встречающееся гетерогенное по этиологии заболевание, проявляющееся в первые 6 месяцев жизни. Различают две основные клинические группы:
- транзито́рный (преходящий) неонатальный сахарный диабет и
- пермане́нтный (персистирующий) неонатальный сахарный диабет.
Неимму́нные фо́рмы са́харного диабе́та у дете́й представляют собой гетерогенную группу различных патологий, характеризующихся этиологическими, патогенетическими и клиническими особенностями. Принятый ISPAD консенсус, определяет сахарный диабет как группу метаболических заболеваний, характеризующихся повышенным уровнем глюкозы в крови, обусловленным нарушением секреции инсулина, механизма действия инсулина либо обеими причинами. Одним словом, нарушения углеводного, жирового и белкового обмена, развивающиеся на фоне сахарного диабета, являются следствием дефицита действия инсулина на ткани-мишени. Данное представление о сахарном диабете нашло отражение в последней версии этиологической классификации Всемирной организации здравоохранения по нарушениям гликемии.

DIDMOAD-синдро́м — аутосомно-рецессивно наследуемый синдром, ассоциированный с инсулинозависимым сахарным диабетом и прогрессирующей атрофией диска зрительного нерва, которые выявляют до 16-летнего возраста. Сочетается с двусторонней прогрессирующей нейросенсорной тугоухостью, несахарным диабетом центрального генеза, дисфункцией автономной нервной системы, приводящей к развитию нейропатического мочевого пузыря и другим проявлениям нейродегенерации, включающими мозжечковую атаксию, миоклональную эпилепсию и атрофию ствола головного мозга. Развёрнутая клиническая картина (фенотипически) встречается приблизительно у 75 % пациентов. Сахарный диабет неаутоиммунного генеза, клинические проявления недостаточности инсулина проявляются приблизительно в 6-летнем возрасте. Средняя продолжительность жизни достигает 30 лет, в течение этого срока происходит развитие полного фенотипа данного синдрома.
Митохондриа́льный са́харный диабе́т — группа генетически обусловленных заболеваний, наследующихся исключительно по материнской линии, проявляющихся сочетанием клинической картины сахарного диабета и сопутствующих неврологических заболеваний: тугоухость, миопатии или неврологические нарушения.
Аутоимму́нный полиэндокри́нный синдро́м — аутоиммунное заболевание, объединяющее поражение нескольких эндокринных органов.
Синдром Макла — Уэльса (MWS) — это мутация в гене CIAS1 с развитием холодового аутовоспалительного синдрома. Является редким аутовоспалительным заболеванием наследственного характера. Преимущественно этническая распространённость — народы Северной Европы. Тип наследования — аутосомно-доминантный. Генетическая основа — мутация гена CIAS1, расположенного на длинном плече 1-й хромосомы (1q44) и кодирующего белка криопирина. Данный белок является основной образуемого в клетке супрамолекулярного комплекса, называемого инфламмасомой, выполняющего функцию превращения pro-IL-1β в активную форму, а также принимающего участие в выполнении программы апоптоза. Холодовой аутовоспалительный синдром тесно связан с двумя другими синдромами: семейной холодовой крапивницы и мультисистемным воспалительным заболеванием неонатального возраста — фактически, все связаны с мутациями гена. В целом Синдром Макла — Уэльса относится к группе криопирин-связанных периодических синдромов (CAPS).
Втори́чные фо́рмы са́харного диабе́та — разнородная группа заболеваний, к которой относится сахарный диабет, встречающийся на фоне другой клинической патологии, которая может и не сочетаться с сахарным диабетом. Для большинства заболеваний из этой группы этиологические факторы раскрыты. Кроме того, к этой группе заболеваний также относят и некоторые генетические (наследственные) синдромы, в том числе аномалии инсулиновых рецепторов. При вторичных формах сахарного диабета отсутствуют ассоциации с HLA-антигенами, данные за аутоиммунное поражение и антитела к островковой ткани поджелудочной железы.
Синдром Ушера — сравнительно редкое генетическое заболевание, вызываемое мутацией одного из 10 генов, приводящее к врождённой нейросенсорной тугоухости и прогрессирующей потере зрения. Одна из основных причин слепоглухоты. В настоящее время неизлечим. Наследуется по аутосомно-рецессивному принципу.
Хроническая прогрессирующая внешняя офтальмоплегия (CPEO), также известная как прогрессирующая внешняя офтальмоплегия (ПЭО), является типом расстройства глаз, характеризующимся медленно прогрессирующей невозможностью перемещения глаз и бровей. Это часто единственная особенность этого митохондриального заболевания, в этом случае термин CPEO может быть использован как диагноз. У других людей, страдающих от митохондриальной болезни, CPEO происходит в рамках синдрома с участием более чем одной части тела, например, синдром Кернс-Сейр. Изредка CPEO может быть вызвана условиями, отличными от митохондриальных заболеваний.
Синдро́м Ке́рнса — Се́йра (англ. Kearns–Sayre syndrome, сокращённо KSS) — митохондриальная миопатия с типичным началом до 20-летнего возраста. KSS является более серьёзным синдромным вариантом хронической прогрессирующей внешней офтальмоплегии, синдром, который характеризуется изолированным поражением мышц, контролирующих движения век и контролирующих движения глаз. Это приводит к птозу и офтальмоплегии соответственно. KSS включает в себя триаду уже описанных CPEO, двустороннюю пигментную ретинопатию и блокаду сердца. Другие области участия могут включать в себя мозжечковую атаксию, проксимальную мышечную слабость, глухоту, сахарный диабет, дефицит гормона роста, гипопаратиреоз или другие эндокринные нарушения. В обоих этих заболеваниях вовлечение мышц может начаться односторонним, но всегда развивается в двусторонний дефицит, который является прогрессирующим.
Синдром Коккейна , также называемый синдром Нил-Дингуолл — редкое аутосомно-рецессивное, нейродегенеративное расстройство, характеризующееся недостатком роста, нарушением развития нервной системы, аномальной чувствительностью к солнечному свету (фотосенсибилизация), заболеваниями глаз и преждевременным старением. Нездоровый вид и неврологические расстройства являются критериями для диагностики, а светочувствительность, нарушения слуха и ненормальные глаза — другие весьма общие черты. Возможны проблемы любого или всех внутренних органов. Это связано с группой расстройств, называемых лейкодистрофия. В основе расстройства лежит дефект механизма репарации ДНК. Интересно, в отличие от других дефектов репарации ДНК, пациенты с CS не предрасположены к раку или инфекции. Синдром Коккейна редок, но это разрушительная болезнь, которая, как правило, приводит к смерти в первом или втором десятилетии жизни. Мутация специфических генов в синдроме Коккейна известна, но широко распространенные эффекты и его отношения с репарацией ДНК еще не очень хорошо исследованы.
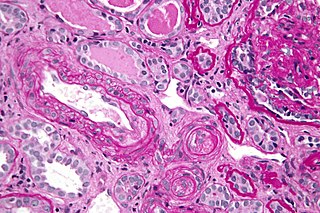
Антифосфолипидный синдром (АФС), или фосфолипидный синдром, или синдром антифосфолипидных антител — аутоиммунное состояние гиперкоагуляции, вызванное антифосфолипидными антителами. АФС провоцирует образование тромбов (тромбоз) как в артериях, так и венах, а также связанные с беременностью осложнения, такие как выкидыш, мертворождение, преждевременные роды и тяжелая преэклампсия.