
Фенилкетонури́я — наследственное заболевание группы ферментопатий, связанное с нарушением метаболизма аминокислот, главным образом фенилаланина. Несоблюдение низкобелковой диеты сопровождается накоплением фенилаланина и его токсических продуктов, что приводит к тяжёлому поражению ЦНС, проявляющемуся, в частности, в виде нарушения умственного развития. Одно из немногих наследственных заболеваний, поддающихся успешному лечению.
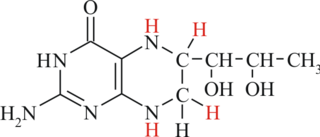
Тетрагидробиоптерин (BH4) — кофермент, участвующий в ряде важных биохимических реакций, в частности в процессах гидроксилирования на этапе промежуточного обмена ароматических аминокислот.
Тирозинемия (тирозиноз) — врождённое заболевание, связанное с дефицитом активности фумарилацетоацетат-гидролазы. «Виновный» ген локализован на 15-й хромосоме: 15q23-q25. Мутации приводят к нарушению метаболизма тирозина с повреждением печени, почек, периферических нервов. Первым пораженным органом является печень, в течение первых месяцев жизни отмечаются начальные проявления печеночной дисфункции с отдаленным исходом в цирроз и печеночную карциному. Как правило, присутствует повреждение тубулярного транспорта с развитием тяжелого рахита ввиду потери фосфатов. У некоторых пациентов развивается нефрокальциноз и почечная недостаточность.
Безглютеновая диета — диета, которая предполагает полное исключение пищи, содержащей клейковину (глютен). Безглютеновая диета является единственным признанным в медицине методом лечения целиакии или связанных с этим заболеванием симптомов. Согласно докладу Всемирной организации гастроэнтерологов (ВОГ-OMGE) за февраль 2005 год, пациенты с целиакией не должны употреблять пшеницу, рожь или ячмень в пищу в каком-либо виде. У пациентов с активной целиакией имеется повышенный риск смерти в сравнении с общей популяцией населения. Однако, этот повышенный риск смертельного исхода возвращается к обычному после трёх-пяти лет строгого соблюдения безглютеновой диеты.
Паренхимато́зные диспротеино́зы — дисметаболические процессы с преимущественным нарушением обмена белков, развивающиеся первично в паренхиматозных клетках органов.

Боле́знь По́мпе — редкое, наследственное заболевание с аутосомно-рецессивным механизмом, наследования,, связанное с повреждением мышечных, и нервных клеток по всему организму. Клиническая картина данной патологии, обусловлена накоплением гликогена, в лизосомах, вызванным недостаточностью лизосомного фермента — кислой α-1,4-глюкозидазы. Различают быстро прогрессирующую (классическую) и медленно прогрессирующую формы болезни Помпе. Несмотря на широкий спектр клинических проявлений, гликогеноза II типа, в основе всех форм болезни лежит, дефицит одного фермента, кодируемого геном GAA.
Болезнь Вольмана — – это редкое аутосомно-рецессивное заболевание лизосомального накопления, вызванное повреждающими мутациями гена LIPA. Возраст начала заболевания и темпы его прогрессирования в значительной степени вариабельны и могут быть связаны с природой, лежащих в основе мутаций. Заболевание у пациентов грудного возраста имеет наиболее быстро прогрессирующее течение с развитием проявлений и симптомов в первые недели жизни; эти пациенты редко доживают до возраста, превышающего 6 месяцев. У детей старшего возраста и взрослых заболевание, обычно, характеризуется определенным сочетанием дислипидемии, гепатомегалии, повышением уровня трансаминаз и микровезикулярным стеатозом в биопсийном материале. У большей части пациентов наблюдается повреждение печени с исходом в фиброз, цирроз и печеночную недостаточность. Частыми изменениями являются повышение уровней холестерина липопротеинов низкой плотности и снижение уровней холестерина липопротеинов высокой плотности. Начиная с детского возраста, могут проявляться и нарушения со стороны сердечно-сосудистой системы. Учитывая, что эти клинические проявления могут наблюдаться и при других сердечно-сосудистых нарушениях, заболеваниях печени и метаболических расстройствах, неудивительно, что LAL-D часто не диагностируется в клинической практике.
Синдро́м Ке́рнса — Се́йра (англ. Kearns–Sayre syndrome, сокращённо KSS) — митохондриальная миопатия с типичным началом до 20-летнего возраста. KSS является более серьёзным синдромным вариантом хронической прогрессирующей внешней офтальмоплегии, синдром, который характеризуется изолированным поражением мышц, контролирующих движения век и контролирующих движения глаз. Это приводит к птозу и офтальмоплегии соответственно. KSS включает в себя триаду уже описанных CPEO, двустороннюю пигментную ретинопатию и блокаду сердца. Другие области участия могут включать в себя мозжечковую атаксию, проксимальную мышечную слабость, глухоту, сахарный диабет, дефицит гормона роста, гипопаратиреоз или другие эндокринные нарушения. В обоих этих заболеваниях вовлечение мышц может начаться односторонним, но всегда развивается в двусторонний дефицит, который является прогрессирующим.

Церебральная фолатная недостаточность — синдром, при котором в спинномозговой жидкости пациента снижено содержание 5-метилтетрагидрофолата (5-MTHF), несмотря на нормальное содержание 5-MTHF в сыворотке крови. Набор симптомов варьирует в зависимости от возраста начала заболевания и его причины, и может включать дискинезию, атаксию, эпилептические приступы, задержку психомоторного развития.

Недостаточность декарбоксилазы ароматических аминокислот - редкое аутосомно-рецессивное генетическое заболевание, вызываемое мутациями гена DDC, кодирующего соответствующий фермент.

Фолатная недостаточность — состояние, при котором в организме снижено содержание фолатов — активных форм фолиевой кислоты. Тяжелая фолатная недостаточность может привести к панцитопении и мегалобластной анемии. Недостаток фолатов во время беременности может привести к дефектам нервной трубки у ребёнка, в том числе к расщеплению позвоночника.
6,7-Дигидроптеридинредуктаза — фермент, катализирующий следующую реакцию:
- 5,6,7,8-тетрагидроптеридин + NAD+ = 6,7-дигидроптеридин + H+ + NADH
Недостаточность дигидроптеридинредуктазы — генетическое расстройство синтеза тетрагидробиоптерина (BH4), вызываемое мутациями гена QDPR. Мутации гена нарушают работу фермента 6,7-дигидроптеридинредуктазы (ДГПР), отвечающего за регенерацию BH4. Тип наследования - аутосомно-рецессивный.

Недостаточность 6-пирувоилтетрагидроптеринсинтазы - редкое заболевание, наследуемое по аутосомно-рецессивному типу. Мутации гена PTS при данном заболевании нарушают одну из реакций в цепочке синтеза тетрагидробиоптерина (BH4), в результате чего у пациента развивается избыток аминокислоты фенилаланина. В свою очередь сбой производства тетрагидробиоптерина, необходимого для выработки нескольких нейромедиаторов, приводит к недостатку этих нейромедиаторов. У пациентов наблюдается целый ряд симптомов, включающий аксиальную гипотонию, задержку развития, когнитивные нарушения, эпилептические приступы, но не ограничивающийся ими. Терапия заболевания заключается в восполнении недостающих нейромедиаторов и мерах, направленных на борьбу с гиперфенилаланинемией.
Недостаточность тетрагидробиоптерина – редкое состояние, при котором пациент страдает от недостатка тетрагидробиоптерина - важного кофермента, необходимого для синтеза моноаминовых нейромедиаторов дофамина и серотонина. Патогенез этого состояния разнообразен: на начало 2021 года было известно шесть редких нейрометаболических заболеваний, нарушающих процесс биосинтеза либо рециклинга BH4. При четырех из этих шести заболеваний у пациента может обнаруживаться повышение концентрации фенилаланина. Терапия в основном заключается в восполнении уровней нейромедиаторов и в коррекции гиперфенилаланинемии.
Аутосомно-рецессивная недостаточность ГТФ-циклогидролазы 1 — заболевание, вызываемое нарушением работы фермента ГТФ-циклогидролаза 1, играющего важную роль в цепочке синтеза тетрагидробиоптерина. Это состояние входит в число шести известных на 2020 год причин недостаточности тетрагидробиоптерина, а также является одной из возможных причин дофа-зависимой дистонии.
Аутосомно-доминантная недостаточность ГТФ-циклогидролазы 1 — заболевание, вызываемое нарушением работы фермента ГТФ-циклогидролаза 1, играющего важную роль в цепочке синтеза тетрагидробиоптерина. Это состояние является одной из нескольких известных причин недостаточности тетрагидробиоптерина, а также самой распространённой причиной дофа-зависимой дистонии. У пациентов наблюдается генерализованная дистония с изменением тяжести симптомов в течение дня, а назначение леводопы приводит к радикальному улучшению состояния.
Недостаточность сепиаптеринредуктазы — заболевание, вызываемое нарушением работы фермента сепиаптеринредуктазы, участвующего в цепочке синтеза тетрагидробиоптерина. Развивающаяся при этом заболевании недостаточность тетрагидробиоптерина вызывает снижение уровней нейромедиатора дофамина, вследствие чего у пациентов отмечаются двигательные нарушения, такие как дистония и окулогирные кризы.
Слабовыраженная гиперфенилаланинемия без дефицита тетрагидробиоптерина — редкое метаболическое заболевание, при котором наблюдается небольшое повышение концентрации фенилаланина в крови, а также ряд неврологических нарушений различного характера и степени тяжести, в том числе двигательные нарушения и расстройства интеллекта. Заболевание наследуется по аутосомно-рецессивному типу. Альтернативное название заболевания — недостаточность DNAJC12.
Синдром гиперфосфатазии с умственной отсталостью – заболевание, при котором у пациентов отмечается умственная отсталость различной степени тяжести и устойчивое повышение активности щелочной фосфатазы в крови. По состоянию на 2020 год было известно шесть генов, мутации которых приводят к развитию синдрома Мабри.