Нейрофармакология
Нейрофармакология — раздел фармакологии, изучающий влияние лекарственных средств на функции нервной системы и регуляцию поведенческих механизмов[1]. Существует два основных направления нейрофармакологии: поведенческая и молекулярная. Поведенческая нейрофармакология фокусируется на том, как лекарственные средства влияют на поведение человека (нейропсихофармакология), а также на изучении влияния зависимости на мозг человека[2]. Молекулярная нейрофармакология включает изучение нейронов и их нейрохимических взаимодействий с целью разработки лекарств, оказывающих благотворное влияние на неврологическую функцию. Обе эти области тесно связаны, поскольку они связаны с нейротрансмиттерами, нейропептидами, нейрогормонами, нейромодуляторами, ферментами, вторичными мессенджерами, котранспортерами, ионными каналами и белками-рецепторами в центральной и периферической нервной системе. Изучая эти взаимодействия, исследователи разрабатывают лекарства для лечения многих неврологических расстройств, включая боль, нейродегенеративные заболевания, такие как болезнь Паркинсона и болезнь Альцгеймера, психические расстройства, зависимости и др.
История
Становление нейрофармакологии как науки начинается с того момента, когда в начале XX века ученые смогли сформулировать базовое понимание нервной системы, а также взаимодействие нервных клеток между собой. В 1930-х годах французские ученые начали работать с соединением под названием фенотиазин, в надежде синтезировать лекарство, способное бороться с малярией. Хотя применение этого препарата было маловероятным у больных малярией, вскоре обнаружили, что он оказывает седативное действие наряду с благотворным действием на пациентов с болезнью Паркинсона. Такие исследования получили название «метод „черного ящика“», что означало введение ученым препарата и изучение его эффекта без понимания связи действия препарата и реакции пациента. Такой метод был основным подходом в этой области, до тех пор пока в конце 1940-х — начале 1950-х годов ученые не идентифицировали специфические нейротрансмиттеры, такие как норэпинефрин, дофамин и серотонин. В 1950-х годах исследователи также стали точнее измерять уровень определённых нейрохимических веществ в организме и таким образом соотносить этот уровень с поведением[3]. Появление метода «зажима напряжения» в 1949 году позволило изучить ионные каналы и потенциал действия нервной клетки. Эти два важнейших исторических события в нейрофармакологии позволили ученым не только изучить, как информация передается от одного нейрона к другому, но и понять, как обрабатывается полученная информация на нейрональном уровне[4].
Обзор
Нейрофармакология — очень широкая область науки, охватывающая многие аспекты нервной системы, от манипуляций с отдельными нейронами до целых областей центральной и периферической нервной системы. Чтобы лучше разобраться в основах разработки лекарственных препаратов, нужно сначала понять, как нейроны взаимодействуют друг с другом.
Нейрохимические взаимодействия
Чтобы оценить потенциальные достижения в медицине, которые может дать нейрофармакология, важно понимать, как человеческое поведение и мыслительные процессы передаются от нейрона к нейрону и как лекарства могут изменить химические основы этих процессов.
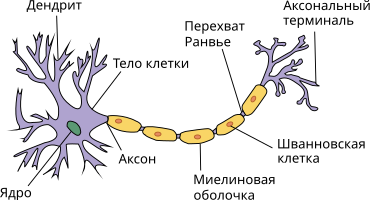
Нейроны известны как возбудимые клетки, потому что на поверхности их мембраны имеется множество ионных каналов, которые позволяют ионам двигаться в обоих направлениях. Структура нейрона позволяет дендритам получать химическую информацию, переносить её через перикарион вниз по аксону и в конечном итоге передавать другим нейронам через терминали аксона. Потенциалозависимые ионные каналы обеспечивают быструю деполяризацию по всей клетке, которая, достигая определённого порога, вызывает потенциал действия. Как только потенциал действия доходит до окончания аксона, он вызывает приток ионов кальция в клетку. Затем ионы кальция заставляют везикулы, заполненные нейротрансмиттерами, связываться с клеточной мембраной и высвобождать её содержимое в синапс. Эта клетка известна как пресинаптический нейрон, а клетка, которая взаимодействует с высвобождаемыми нейротрансмиттерами, — постсинаптический нейрон. Как только нейротрансмиттер высвобождается в синапс, он может либо связываться с рецепторами постсинаптической клетки, либо захватываться обратно пресинаптической клеткой и сохраняться для последующей передачи. Также нейротрансмиттеры могут расщепляться специфическими ферментами в синапсе. Это три разных пути, в процессе которых действие лекарственных средств может влиять на связь между нейронами[3].
Есть два типа рецепторов, с которыми нейротрансмиттеры взаимодействуют на постсинаптических нейронах. Первыми типами рецепторов являются лиганд-управляемые ионные каналы, или LGIC. Рецепторы LGIC самые быстрые по преобразованию химического сигнала в электрический. Как только нейротрансмиттер связывается с рецептором, он вызывает конформационные изменения, которые позволяют ионам напрямую проникать в клетку. Второй тип известен как рецепторы, связанные с G-белком, или GPCR. Они намного медленнее, чем LGIC, из-за большего количества биохимических реакций, которые должны происходить внутри клетки. Как только нейротрансмиттер связывается с белком GPCR, он вызывает каскад внутриклеточных взаимодействий, которые могут привести к множеству различных изменений в клеточной биохимии, физиологии и экспрессии генов. Взаимодействия «нейротрансмиттер — рецептор» в области нейрофармакологии чрезвычайно важны, поскольку многие лекарства, которые разрабатываются сегодня, связаны с нарушением этого процесса[5].
Молекулярная нейрофармакология
Молекулярная нейрофармакология включает изучение нейронов и их нейрохимические взаимодействия, а также рецепторов с целью разработки новых лекарств, которые будут применяться при лечении неврологических расстройств, таких как боль, нейродегенеративные заболевания и психологические расстройства. Есть несколько основных определений, понимание которых связывает нейротрансмиссию с действием рецепторов:
1) агонист — молекула, которая связывается с рецепторным белком и активирует этот рецептор;
2) конкурентный антагонист — молекула, которая связывается с тем же участком на рецепторном белке, что и агонист, предотвращая активацию рецептора;
3) неконкурентный антагонист — молекула, которая связывается с белком-рецептором на сайте, отличном от сайта агониста, но вызывает конформационное изменение белка, не допускающее активации.
На взаимодействие «нейротрансмиттер — рецептор» могут влиять синтетические соединения, которые действуют как одно из трех вышеперечисленных. Натрий-калиевые каналы также могут манипулировать нейроном, чтобы вызвать ингибирующий эффект потенциала действия.
Гамма-аминомасляная кислота (ГАМК)
Нейротрансмиттер ГАМК опосредует быстрое синаптическое торможение в центральной нервной системе. Когда ГАМК высвобождается из пресинаптической клетки, она связывается с рецептором (скорее всего, рецептором ГАМКА), что вызывает гиперполяризацию постсинаптической клетки. Это противодействует эффекту возбуждения.
Рецептор ГАМКА содержит множество сайтов связывания, которые допускают конформационные изменения и являются основной мишенью для разработки лекарственных препаратов. Наиболее распространенный из этих сайтов связывания, бензодиазепин, позволяет оказывать как агонистическое, так и антагонистическое действие на рецептор. Распространенный препарат диазепам действует как аллостерический усилитель в этом месте связывания[6]. Другой рецептор ГАМК, известный как ГАМКB, может быть активирован молекулой, называемой баклофен. Данное соединение действует как агонист, активируя рецептор, и помогает контролировать гипертонус мышц.
Дофамин
Дофамин опосредует синаптическую передачу, связываясь с пятью специфическими GPCR. Эти пять рецепторных белков разделены на два класса в зависимости от того, вызывает их активация возбуждающий или тормозной ответ постсинаптической клетки. Есть много видов веществ, легальных и нелегальных, которые влияют на дофамин и его взаимодействие в мозге. При болезни Паркинсона, заболевании, которое снижает количество дофамина в головном мозге, пациенту назначают предшественник дофамина леводопу, который может проникать через гематоэнцефалический барьер. Некоторые агонисты дофамина также назначаются пациентам с болезнью Паркинсона, у которых есть расстройство, известное как синдром беспокойных ног. Некоторыми примерами из них являются ропинирол и прамипексол[7].
Психологические расстройства, такие как синдром дефицита внимания и гиперактивности (СДВГ), можно лечить с помощью препарата метилфенидат (известный как риталин), который блокирует повторное поглощение дофамина пресинаптической клеткой, тем самым обеспечивая увеличение дофамина, оставшегося в синаптической щели. Это увеличение синаптического дофамина повышает вероятность связывания с рецепторами постсинаптической клетки. Этот же механизм используется нелегальными и более сильнодействующими стимуляторами, такими как кокаин.
Серотонин
Серотонин обладает способностью опосредовать синаптическую передачу через рецепторы GPCR или LGIC. Возбуждающие или тормозящие постсинаптические эффекты серотонина определяются типом рецептора, экспрессируемого в данной области мозга. Наиболее популярные и широко используемые препараты для регуляции серотонина при депрессии — селективные ингибиторы обратного захвата серотонина (СИОЗС). Эти препараты ингибируют захват серотонина обратно в пресинаптический нейрон, оставляя больше медиатора в синаптической щели.
До открытия СИОЗС также существовали препараты, ингибирующие фермент, расщепляющий серотонин. Ингибиторы моноаминоксидазы (ИМАО) увеличивали количество серотонина в синапсах, но имели много побочных эффектов, включая сильную мигрень и повышенное кровяное давление. В конечном итоге это было связано с лекарствами, взаимодействующими с обычным химическим веществом, известным как тирамин, который содержится во многих видах пищи, например в сыре[8].
Ионные каналы
Ионные каналы, расположенные на поверхности мембраны нейрона, обеспечивают транспорт ионов натрия и калия в клетку и из неё. Избирательная блокировка этих ионных каналов снижает вероятность возникновения потенциала действия. Препарат рилузол является нейропротектором, блокирующим натриевые ионные каналы. Поскольку эти каналы не могут активироваться в отсутствие потенциала действия, нейрон не выполняет никакого преобразования химических сигналов в электрические и сигнал не движется дальше. Этот препарат используется в качестве анестетика, а также седативного средства[9].
Поведенческая нейрофармакология

Одна из форм поведенческой нейрофармакологии фокусируется на изучении наркотической зависимости и её влияния на человеческий разум. Большинство исследований показало, что основной частью мозга, усиливающей зависимость посредством нейрохимического вознаграждения, является прилежащее ядро. На изображении справа показано, как дофамин проецируется в эту область. Длительное чрезмерное употребление алкоголя может вызвать зависимость и привыкание.
Этанол
Вознаграждающие и подкрепляющие (то есть вызывающие привыкание) свойства алкоголя опосредованы его воздействием на дофаминовые нейроны в мезолимбическом пути вознаграждения, который соединяет вентральную область покрышки с прилежащим ядром (NAcc)[10][11]. Одним из первичных эффектов алкоголя является аллостерическое ингибирование рецепторов NMDA и содействие активации рецепторов ГАМКА (например, усиленный поток хлоридов, опосредованный рецептором ГАМКА посредством аллостерической регуляции рецептора)[12]. В высоких дозах этанол ингибирует большинство лигандзависимых ионных каналов, а также потенциалзависимых ионных каналов в нейронах[12]. Алкоголь подавляет натриево-калиевые насосы в мозжечке, вероятно, именно поэтому ухудшая его работу и, как следствие, координацию тела[13][14].
При остром отравлении алкоголем в синапсах мезолимбического пути высвобождается дофамин, что, в свою очередь, усиливает активацию постсинаптических D1-рецепторов[10][11]. Активация этих рецепторов запускает постсинаптические внутренние сигнальные события через протеинкиназу А, которые в конечном итоге фосфорилируют белок, связывающий элемент ответа цАМФ (CREB), вызывая CREB-опосредованные изменения в экспрессии генов[10][11].
При хроническом алкоголизме потребление этанола индуцирует фосфорилирование CREB через путь рецептора D1, а также изменяет функцию рецептора NMDA посредством механизмов фосфорилирования[10][11]. Хроническое потребление также связано с влиянием на фосфорилирование и функцию CREB посредством сигнальных каскадов постсинаптических рецепторов NMDA через путь MAPK/ERK и CAMK-опосредованный путь[11]. Эти модификации функции CREB в мезолимбическом пути индуцируют экспрессию (то есть увеличение экспрессии генов) ΔFosB в NAcc[11], где ΔFosB является «главным контрольным белком», который при сверхэкспрессии в NAcc необходим и достаточен для развития и поддержания аддиктивного состояния (то есть его сверхэкспрессия в прилежащем ядре вызывает, а затем напрямую модулирует компульсивное потребление алкоголя)[11][15][16][17].
Исследования
Болезнь Паркинсона
Болезнь Паркинсона — нейродегенеративное заболевание, характеризующееся избирательной гибелью дофаминергических нейронов, расположенных в черной субстанции. Сегодня наиболее часто используемым препаратом для лечения является леводопа, или L-ДОФА. Этот предшественник дофамина может проникать через гематоэнцефалический барьер, тогда как дофамин не может. Было проведено обширное исследование, чтобы определить, является ли L-ДОФА лучшим средством для лечения болезни Паркинсона, чем другие агонисты дофамина. Некоторые считают, что длительное использование леводопы нарушает нейрозащиту и в конечном итоге приводит к гибели дофаминергических клеток. Хотя доказательств этому не было in vivo или in vitro, есть мнение, что долгосрочное использование агонистов дофамина безопаснее для пациентов[18].
Болезнь Альцгеймера
Несмотря на то что существует множество гипотез, которые были предложены в качестве причин болезни Альцгеймера, знания об этом заболевании далеки от полного объяснения, что затрудняет разработку методов лечения. Известно, что при данном заболевании подавляются как нейрональные никотиновые ацетилхолиновые (nACh) рецепторы, так и рецепторы NMDA. В США были разработаны и одобрены Управлением по продуктам питания и лекарствам США (FDA) четыре антихолинэстеразных препарата. Однако они не являются оптимальными, учитывая их побочные эффекты и ограниченную эффективность. Один препарат, нефирацетам, разрабатываемый для лечения пациентов с болезнью Альцгеймера и с деменцией, обладает уникальным действием по усилению активности как рецепторов nACh, так и NMDA-рецепторов[19].
Будущее
Благодаря достижениям в области технологий, разработка лекарств будет продолжаться в направлении повышения чувствительности и специфичности лекарств. Связь «структура — активность» является основной областью исследований в нейрофармакологии; предпринимаются попытки модифицировать эффект или активность биоактивных химических соединений путем изменения их химической структуры[9].
Примечания
- ↑ Yeung AWK, Tzvetkov NT, Atanasov AG. When Neuroscience Meets Pharmacology: A Neuropharmacology Literature Analysis Архивная копия от 16 декабря 2018 на Wayback Machine. Front Neurosci. 2018 Nov 16;12:852. doi: 10.3389/fnins.2018.00852 Архивная копия от 16 декабря 2018 на Wayback Machine.
- ↑ Everitt, B. J. (2005). "Neural systems of reinforcement for drug addiction: from actions to habits to compulsion". Nature Neuroscience. 8 (11): 1481—1489. doi:10.1038/nn1579. PMID 16251991.
- ↑ 1 2 Wrobel, S. (2007). "Science, serotonin, and sadness: the biology of antidepressants: A series for the public". The FASEB Journal. 21 (13): 3404—17. doi:10.1096/fj.07-1102ufm. PMID 17967927.
{{cite journal}}: Википедия:Обслуживание CS1 (не помеченный открытым DOI) (ссылка) - ↑ Andrew Huxley. From overshoot to voltage clamp (англ.) // Trends in Neurosciences. — 2002-11. — Vol. 25, iss. 11. — P. 553–558. — doi:10.1016/S0166-2236(02)02280-4. Архивировано 22 мая 2023 года.
- ↑ Lovinger, D. M. (2008). "Communication Networks in the Brain Neurons, Receptors, Neurotransmitters, and Alcohol. [Review]". Alcohol Research & Health. 31 (3): 196—214.
- ↑ Sigel, E (2002). "Mapping of the benzodiazepine recognition site on GABA(A) receptors". Current Topics in Medicinal Chemistry. 2 (8): 833—9. doi:10.2174/1568026023393444. PMID 12171574.
- ↑ Winkelman, JW (2007). "Restless legs syndrome: nonpharmacologic and pharmacologic treatments". Geriatrics. 62 (10): 13—6. PMID 17922563.
- ↑ López-Muñoz, F. (2009). "Monoaminergic neurotransmission: the history of the discovery of antidepressants from 1950s until today". Current Pharmaceutical Design. 15 (14): 1563—1586. doi:10.2174/138161209788168001. PMID 19442174.
- ↑ 1 2 Narahashi, T (2000). "Neuroreceptors and ion channels as the basis for drug action: past, present, and future". The Journal of Pharmacology and Experimental Therapeutics. 294 (1): 1—26. PMID 10871290.
- ↑ 1 2 3 4 Alcoholism – Homo sapiens (human) Database entry. KEGG Pathway (29 октября 2014). — «As one of the primary mediators of the rewarding effects of alcohol, dopaminergic ventral tegmental area (VTA) projections to the nucleus accumbens (NAc) have been identified. Acute exposure to alcohol stimulates dopamine release into the NAc, which activates D1 receptors, stimulating PKA signaling and subsequent CREB-mediated gene expression, whereas chronic alcohol exposure leads to an adaptive downregulation of this pathway, in particular of CREB function. The decreased CREB function in the NAc may promote the intake of drugs of abuse to achieve an increase in reward and thus may be involved in the regulation of positive affective states of addiction. PKA signaling also affects NMDA receptor activity and may play an important role in neuroadaptation in response to chronic alcohol exposure.» Дата обращения: 9 февраля 2015. Архивировано 11 августа 2019 года.
- ↑ 1 2 3 4 5 6 7 Kanehisa Laboratories. Alcoholism – Homo sapiens (human). KEGG Pathway (29 октября 2014). Дата обращения: 31 октября 2014. Архивировано 12 ноября 2019 года.
- ↑ 1 2 Chapter 15: Reinforcement and Addictive Disorders // Molecular Neuropharmacology: A Foundation for Clinical Neuroscience. — 2nd. — New York : McGraw-Hill Medical, 2009. — P. 372. — «Despite the high concentrations required for its psychoactive effects, ethanol exerts specific actions on the brain. The initial effects of ethanol result primarily from facilitation of GABAA receptors and inhibition of NMDA glutamate receptors. At higher doses, ethanol also inhibits the functioning of most ligand- and voltage-gated ion channels. It is not known whether ethanol selectively affects these channels via direct low affinity binding or via nonspecific disruption of plasma membranes which then selectively influences these highly complex, multimeric, transmembrane proteins. Ethanol allosterically regulates the GABAA receptor to enhance GABA-activated Cl− flux. The anxiolytic and sedative effects of ethanol, as well as those of barbiturates and benzodiazepines, result from enhancement of GABAergic function. Facilitation of GABAA receptor function is also believed to contribute to the reinforcing effects of these drugs. Not all GABAA receptors are ethanol sensitive. ... Ethanol also acts as an NMDA antagonist by allosterically inhibiting the passage of glutamate-activated Na+ and Ca2+ currents through the NMDA receptor. ... The reinforcing effects of ethanol are partly explained by its ability to activate mesolimbic dopamine circuitry, although it is not known whether this effect is mediated at the level of the VTA or NAc. It also is not known whether this activation of dopamine systems is caused primarily by facilitation of GABAA receptors or inhibition of NMDA receptors, or both. Ethanol reinforcement also is mediated in part by ethanol-induced release of endogenous opioid peptides within the mesolimbic dopamine system, although whether the VTA or NAc is the predominant site of such action is not yet known. Accordingly, the opioid receptor antagonist naltrexone reduces ethanol self-administration in animals and is used with modest effect to treat alcoholism in humans.». — ISBN 9780071481274.
- ↑ Forrest MD (April 2015). "Simulation of alcohol action upon a detailed Purkinje neuron model and a simpler surrogate model that runs >400 times faster". BMC Neuroscience. 16 (27): 27. doi:10.1186/s12868-015-0162-6. PMID 25928094.
{{cite journal}}: Википедия:Обслуживание CS1 (не помеченный открытым DOI) (ссылка) - ↑ Forrest. The neuroscience reason we fall over when drunk. Science 2.0 (апрель 2015). Дата обращения: 2 января 2019. Архивировано 25 августа 2017 года.
- ↑ Ruffle JK (November 2014). "Molecular neurobiology of addiction: what's all the (Δ)FosB about?". Am J Drug Alcohol Abuse. 40 (6): 428—437. doi:10.3109/00952990.2014.933840. PMID 25083822.
ΔFosB as a therapeutic biomarker
The strong correlation between chronic drug exposure and ΔFosB provides novel opportunities for targeted therapies in addiction (118), and suggests methods to analyze their efficacy (119). Over the past two decades, research has progressed from identifying ΔFosB induction to investigating its subsequent action (38). It is likely that ΔFosB research will now progress into a new era – the use of ΔFosB as a biomarker. If ΔFosB detection is indicative of chronic drug exposure (and is at least partly responsible for dependence of the substance), then its monitoring for therapeutic efficacy in interventional studies is a suitable biomarker (Figure 2). Examples of therapeutic avenues are discussed herein. ...
Conclusions
ΔFosB is an essential transcription factor implicated in the molecular and behavioral pathways of addiction following repeated drug exposure. The formation of ΔFosB in multiple brain regions, and the molecular pathway leading to the formation of AP-1 complexes is well understood. The establishment of a functional purpose for ΔFosB has allowed further determination as to some of the key aspects of its molecular cascades, involving effectors such as GluR2 (87,88), Cdk5 (93) and NFkB (100). Moreover, many of these molecular changes identified are now directly linked to the structural, physiological and behavioral changes observed following chronic drug exposure (60,95,97,102). New frontiers of research investigating the molecular roles of ΔFosB have been opened by epigenetic studies, and recent advances have illustrated the role of ΔFosB acting on DNA and histones, truly as a molecular switch (34). As a consequence of our improved understanding of ΔFosB in addiction, it is possible to evaluate the addictive potential of current medications (119), as well as use it as a biomarker for assessing the efficacy of therapeutic interventions (121,122,124). Some of these proposed interventions have limitations (125) or are in their infancy (75). However, it is hoped that some of these preliminary findings may lead to innovative treatments, which are much needed in addiction. - ↑ Nestler EJ (December 2013). "Cellular basis of memory for addiction". Dialogues Clin Neurosci. 15 (4): 431—443. PMID 24459410.
DESPITE THE IMPORTANCE OF NUMEROUS PSYCHOSOCIAL FACTORS, AT ITS CORE, DRUG ADDICTION INVOLVES A BIOLOGICAL PROCESS: the ability of repeated exposure to a drug of abuse to induce changes in a vulnerable brain that drive the compulsive seeking and taking of drugs, and loss of control over drug use, that define a state of addiction. ... A large body of literature has demonstrated that such ΔFosB induction in D1-type NAc neurons increases an animal's sensitivity to drug as well as natural rewards and promotes drug self-administration, presumably through a process of positive reinforcement
- ↑ "Transcriptional and epigenetic mechanisms of addiction". Nat. Rev. Neurosci. 12 (11): 623—637. November 2011. doi:10.1038/nrn3111. PMID 21989194.
ΔFosB has been linked directly to several addiction-related behaviors ... Importantly, genetic or viral overexpression of ΔJunD, a dominant negative mutant of JunD which antagonizes ΔFosB- and other AP-1-mediated transcriptional activity, in the NAc or OFC blocks these key effects of drug exposure14,22–24. This indicates that ΔFosB is both necessary and sufficient for many of the changes wrought in the brain by chronic drug exposure. ΔFosB is also induced in D1-type NAc MSNs by chronic consumption of several natural rewards, including sucrose, high fat food, sex, wheel running, where it promotes that consumption14,26–30. This implicates ΔFosB in the regulation of natural rewards under normal conditions and perhaps during pathological addictive-like states. ... ΔFosB serves as one of the master control proteins governing this structural plasticity.
- ↑ Shin, J. Y. (2009). "Neuroprotective effect of l-dopa on dopaminergic neurons is comparable to pramipexol in MPTP-treated animal model of Parkinson's disease: a direct comparison study". Journal of Neurochemistry. 111 (4): 1042—50. doi:10.1111/j.1471-4159.2009.06381.x. PMID 19765187.
- ↑ Narahashi, T (2003). "Unique mechanism of action of Alzheimer's drugs on brain nicotinic acetylcholine receptors and NMDA receptors". Life Sciences. 74 (2—3): 281—91. doi:10.1016/j.lfs.2003.09.015. PMID 14607256.
Ссылки
 На Викискладе есть медиафайлы по теме Нейрофармакология
На Викискладе есть медиафайлы по теме Нейрофармакология
| Лиганд (биохимия) |
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Фармакодинамика |
| ||||||||
| Фармакокинетика |
| ||||||||
| Смежные области |
| ||||||||
| Другое |
| ||||||||