Нейрофиброматоз I типа
| Нейрофиброматоз I типа | |
|---|---|
 «Кофейные пятна» на коже. | |
| МКБ-11 | LD2D.10 |
| МКБ-10 | Q85.0 |
| МКБ-9 | 237.71 |
| OMIM | 162200 |
| DiseasesDB | 8937 |
| MedlinePlus | 000847 |
| eMedicine | derm/287 neuro/248 oph/338 radio/474 |
| MeSH | D009456 |
Нейрофиброматоз I (первого) типа (нейрофиброматоз с феохромоцитомой, болезнь фон Реклингхаузена, синдром Реклингхаузена, NF-1) — самое распространённое наследственное заболевание, предрасполагающее к возникновению опухолей у человека. Описан во второй половине XIX века рядом исследователей, в том числе в 1882 году учеником Рудольфа Вирхова Фридрихом фон Реклингхаузеном. Устаревшие названия — болезнь Реклингхаузена, периферический нейрофиброматоз и др. Является аутосомно-доминантным, встречается с одинаковой частотой у мужчин и у женщин, у 1 из 3500 новорождённых[1][2][3]. Другие типы нейрофиброматозов (на первую половину 2011 года выделяют 7 типов[4][5], из которых наибольшее клиническое значение имеют первые два) характеризуются наличием как сходных проявлений с I типом, так и отличий.
В половине случаев заболевание является наследственным, в половине — результатом спонтанной мутации. Частота мутаций генов, повреждения которых приводит к нейрофиброматозу I типа, является самой высокой из известных для генов человека[1].
Для заболевания характерно появление множественных пигментированных пятен цвета «кофе с молоком», доброкачественных новообразований — нейрофибром, опухолей центральной нервной системы, костных аномалий, изменений радужной оболочки глаза и целого ряда других симптомов.
История
Первое научное описание клинических и морфологических изменений данного заболевания было сделано в 1882 году немецким патологоанатомом Фридрихом фон Реклингхаузеном. Описание заболевания было сделано задолго до открытия структуры ДНК. В связи с этим оно получило название «болезни Реклингхаузена». Данным термином обозначали не только нейрофиброматоз I типа, но и нейрофиброматоз вообще. Различные по причинам возникновения и некоторым проявлениям нейрофиброматоз I и II типа определяли как «периферическую» и «центральную» формы[6][7] болезни Реклингхаузена. После определения генетических причин возникновения термин стал устаревать[8]. В современной медицинской литературе болезнь называется «нейрофиброматоз I типа».
Долгое время считалось, что известный своими уродствами Джозеф Меррик по прозвищу «человек-слон» был болен нейрофиброматозом I типа. Известный фильм Дэвида Линча «Человек-слон» о тяжёлой судьбе Джона Меррика способствовал возникновению в обществе ложного мнения о том, что люди с данным заболеванием имеют ужасную внешность. Современные исследователи предполагают наличие у Меррика синдрома Протея[9][10].
Эпидемиология

Заболеваемость нейрофиброматозом I типа составляет 30—40 больных на 100 тысяч населения, что соответствует одному человеку на 2500—3000. Нейрофиброматоз I типа является аутосомно-доминантным заболеванием с высокой пенетрантностью и высокой частотой возникновения новых мутаций. Риск рождения ребёнка с нейрофиброматозом от больного человека составляет либо 50 % (в случае гетерозиготы) либо 100 % (в случае гомозиготы). Примерно 50 % случаев заболевания представляют мутации лат. de novo[11]. Частота заболеваемости нейрофиброматозом I типа не отличается в разных географических регионах и среди этнических групп[12].
Этиопатогенез
Нейрофиброматоз I типа был первым опухолевым заболеванием с доказанным генетическим происхождением[13]. Локус генов, поломка которых приводит к развитию нейрофиброматоза, располагается на длинном плече 17 хромосомы (17q11.2)[14][15]. Он состоит из 400 тысяч нуклеотидных пар. В нём содержится информация, ответственная за синтез одного из составляющих миелин гликопротеина, нейрофибромина и других белков. При нейрофиброматозе I типа в данном локусе отмечены различные типы мутаций и перестроек — транслокации, делеции, инверсии и точковые мутации[16]. Характер мутаций весьма специфичен: более 80 % из них ведут к синтезу нефункционального «усечённого» белка либо к полному отсутствию транскрипта (нонсенс-мутации, мутации в сайтах сплайсинга, делеции и инсерции со сдвигом рамки, крупные делеции, охватывающие весь ген или его значительную часть)[11][17][18][19][20][21].
Нейрофибромин представляет собой цитоплазматический белок, состоящий из 2818 аминокислот[22]. Он участвует в инактивации белков-промоторов (белка RAS и его аналогов)[23], обеспечивая динамический контроль клеточного роста. Ген НФ-1 является одним из основных генов-супрессоров опухолевого роста для примерно половины тканей организма, в первую очередь нейроэктодермального происхождения, пролиферация которых определяется системой белков RAS[1]. Нейрофибромин также влияет на содержание в клетке аденозинмонофосфата (АМФ). АМФ в свою очередь опосредованно тормозит процессы клеточного деления[22].
При повреждении гена НФ1 в одной из хромосом 17 пары половина синтезируемого нейрофибромина становится дефектной, и отмечается смещение равновесия роста клеток в сторону пролиферации. Остающийся неповреждённым аллельный (находящийся в парной хромосоме) ген НФ1 обеспечивает синтез нормального нейрофибромина. Выраженность клинических проявлений нейрофиброматоза определяется состоянием общего противоопухолевого иммунитета и может варьировать в широких пределах. Возникают доброкачественные новообразования[1].
В случае утраты вследствие мутации аллельного нормального гена НФ1 возникает бурный неконтролируемый рост клеток, то есть развивается злокачественная опухоль (чаще всего нейрофибросаркома или нейробластома)[1]. Вероятность их возникновения составляет 3—15 %[24]. Вероятность развития ассоциированной с нейрофиброматозом I типа злокачественной опухоли превышает таковую в популяции в сотни раз (только в отношении миелолейкоза в 200—500 раз)[1].
Клиническая картина


Нейрофиброматоз I типа проявляется рядом патогномоничных симптомов. К ним относят наличие пигментных пятен на коже цвета «кофе с молоком», нейрофибром, большинство из которых располагаются поверхностно на коже, узелки Лиша — гамартомы радужной оболочки глаза[25].
Проявления нейрофиброматоза I типа часто начинаются со сколиоза (искривления позвоночника), затем возникают трудности в обучении, проблемы со зрением и эпилепсия.
Нейрофибромы чаще локализуются по ходу периферических нервов. Однако может поражаться спинной и головной мозг, находят нейрофибромы на веках, конъюнктиве, в средостении, брюшной полости. В зависимости от расположения нейрофибромы могут вызвать различную клиническую симптоматику: судороги, нарушение функции черепных нервов и сегментов спинного мозга, паралич глазных мышц, птоз, сдавление органов средостения.
Нейрофибромы

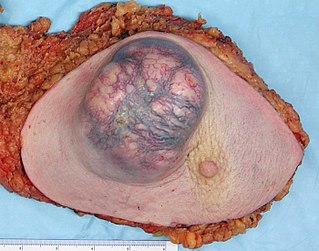
Для данного заболевания характерно появление большого количества нейрофибром, как кожных, так и плексиформных. Кожные нейрофибромы представлены небольшими доброкачественными и ограниченными новообразованиями. Они располагаются подкожно, растут на оболочках мелких нервов кожи. Плексиформные нейрофибромы развиваются на крупных нервах и приводят к нарушению их функций[26]. Также плексиформные нейрофибромы характеризуются своими большими размерами. Встречаются у 30 % больных нейрофиброматозом I типа[22].
Клинически повреждение нерва проявляется хроническими болями, онемением и/или параличами мышц.
Опухоли центральной нервной системы

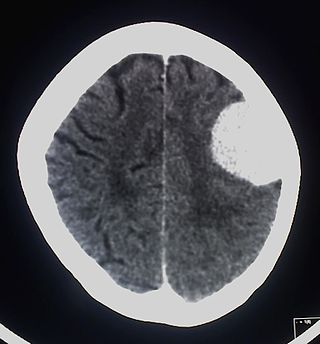
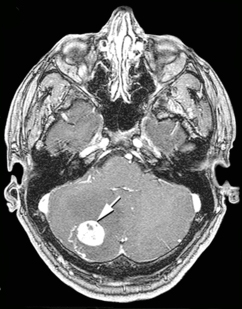
При нейрофиброматозе I типа частота развития опухолей центральной нервной системы составляет от 5[27] до 30 %[28][29]. Во многих случаях опухоли ЦНС у больных нейрофиброматозом не выявляются[30]. Впервые[30] взаимосвязь между нейрофиброматозом I типа и внутричерепными новообразованиями была отмечена в 1940 году[31].
Наиболее часто возникающими при данном заболевании опухолями ЦНС являются глиомы зрительных нервов, астроцитомы, эпендимомы, невриномы слухового нерва, менингиомы и нейрофибромы[32][33][34][35].
Клиническая картина при опухолях ЦНС будет зависеть от их размеров, месторасположения и вовлечённых в патологический процесс образований.
Пигментные нарушения
Для нейрофиброматоза патогномоничны пигментные пятна от светло-бежевого до тёмно-коричневого цвета, которые выявляют на коже туловища и конечностей, реже на лице, шее, слизистой оболочке полости рта. Они имеют гладкую поверхность, не выступают над уровнем кожи. При гистологическом исследовании пигментных пятен обнаруживают диффузное скопление в сосочковом слое дермы меланобластов и меланоцитов с включениями меланина в цитоплазме[36].
Данные пигментные пятна носят характер пятен цвета «кофе с молоком» (фр. café-au-lait, англ. milk coffee) и «веснушчатых гроздьев»[24]. В некоторых случаях пятна имеют синий или фиолетовый цвет, реже встречается депигментация[37].
Узелки Лиша

Узелки Лиша встречаются практически у всех больных нейрофиброматозом I типа старше 20 лет. Они представляют небольшие белесоватые пятна (гамартомы) на радужке глаза. Узелки Лиша не видны невооружённым взглядом, необходимо офтальмологическое обследование. Выявляемость узелков Лиша повышается с возрастом больного: в возрасте от 0 до 4 лет — до 22 % случаев; 5—9 лет — до 41; 10—19 лет — до 85 %; старше 20 лет — до 95 % больных нейрофиброматозом I типа[24]. Данные узелки не встречаются при других формах нейрофиброматоза[37].
Впервые гамартомы радужной оболочки были описаны в 1918 году[38]. Их значение в диагностике нейрофиброматоза I типа было показано в 1937 году австрийским офтальмологом Карлом Лишем, в честь которого они и получили своё название. Впоследствии была установлена их чрезвычайная роль в дифференциальной диагностике болезни Реклингхаузена[39][40].
Костные изменения

Для выраженного нейрофиброматоза характерна деформация позвоночника в виде сколиоза, возможны краевые дефекты тел позвонков, их суставных и поперечных отростков, расширение межпозвоночных отверстий и эрозии их краёв, узуры нижних краёв задних отделов рёбер, вызванные давлением нейрофиброматозных узлов[36].
Длинные трубчатые кости могут быть атрофичными, изогнутыми, иногда же, наоборот, гипертрофированными, утолщёнными. Компактное вещество в гипертрофированной кости утолщено. На поверхности кости видны периостальные гребни, иногда обнаруживаются и параостальные окостенения. Внутрикостные нейрофибромы в трубчатых костях выглядят как ограниченные вздутия и кистовидные образования[36].
При вовлечении в процесс костей черепа обнаруживается его асимметрия. Особенно выраженной она бывает при деформациях его лицевой части и стенок глазницы. В костях свода черепа возможны дефекты и узуры, участки атрофии кости или явления гиперостоза[36].
Дополнительные клинические проявления нейрофиброматоза I типа
- Когнитивные нарушения — затруднения освоения письма, чтения, математики. Часто сочетаются с умеренным снижением интеллекта (IQ<70)[24]. У больных часто отмечаются депрессия из-за стыда, вызываемого обезображиванием тела и лица нейрофибромами, боязнь общества;
- Эндокринные расстройства — феохромоцитома[41], нарушение роста и полового созревания[24];
- Эпилептические припадки[24];
- Снижение мышечного тонуса[24];
- Нарушения поведения[24];
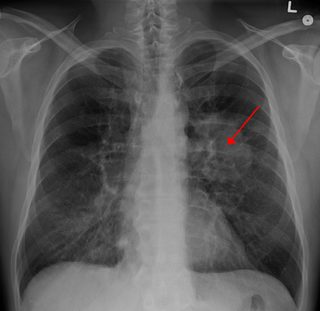
- Заболевания, напрямую не связанные с вовлечением нервов, — стеноз почечной и легочной артерий, легочные кисты, интерстициальная пневмония, гипертрофия клитора, неправильное формирование отделов желудочно-кишечного тракта[1]. Стеноз почечной артерии, особенно в сочетании с феохромоцитомой, обуславливает развитие артериальной гипертензии
- Сирингомиелия[1]


Диагностика
Диагноз нейрофиброматоза I типа может быть поставлен при наличии у больного сочетания двух и более патогномоничных для заболевания симптомов[1][42][43]:
- шесть и более пятен цвета «кофе с молоком» диаметром свыше 5 мм у детей в препубертатном периоде и свыше 15 мм — в постпубертатном;
- наличие двух и более обычных нейрофибром, либо одной плексиформной нейрофибромы;
- гиперпигментация (по типу «веснушчатых гроздьев»[22]) подмышечной и/или паховой области;
- глиомы зрительных нервов;
- два и более узелка Лиша;
- костные аномалии (истончение кортикального слоя трубчатых костей, часто приводящего к формированию ложных суставов, дисплазии крыльев клиновидной кости);
- наличие нейрофиброматоза I типа у ближайших родственников.
Первичная диагностика заболевания производится как врачами общей практики, так и узкими специалистами (неврологами, дерматологами, офтальмологами, нейрохирургами, стоматологами и др.). Процесс развития клинической симптоматики при нейрофиброматозе I типа динамический[24].
| Симптом | Ранний детский возраст (0—2 года) | Дошкольный возраст | Школьники и подростки (6—16 лет) | Взрослые (старше 16 лет) |
|---|---|---|---|---|
| Пятна «кофе с молоком» | ||||
| Диффузные плексиформные нейрофибромы | ||||
| Гиперпигментация подмышечной и/или паховой области | ||||
| Глиомы зрительных нервов | ||||
| Нарушения обучения | ||||
| Артериальная гипертензия | ||||
| Головная боль | ||||
| Кожные нейрофибромы | ||||
| Сколиоз | ||||
| Злокачественные опухоли ЦНС |
Дифференциальная диагностика
Более чем при 100 наследственных болезнях и синдромах выявляется один из основных симптомов нейрофиброматоза I типа — пятна «цвета кофе с молоком» на коже[24].
Чаще всего дифференциальная диагностика проводится с нейрофиброматозом II типа. Возникающие при данном заболевании опухоли являются доброкачественными, но более агрессивными, чем при I типе. Абсолютным диагностическим критерием является наличие у больного двусторонних неврином VIII пары черепных нервов. При II типе возможны и другие внутричерепные новообразования: менингиомы, глиомы, шванномы[44]. Кроме данного заболевания дифференциальная диагностика проводится также с синдромом Протея, синдромом Клиппеля-Тренаунау-Вебера[англ.], рассеянным липоматозом и другими[24].
Лечение
Лечение оперативное. Показаниями для него являются резкая болезненность или изъязвление опухоли, затруднение движений, сдавление или смещение жизненно важных органов. В некоторых случаях к операции прибегают с косметической целью. Так как поражения при нейрофиброматозе множественные, то удаление всех патологических очагов, в большинстве случаев, не представляется возможным[36].
При оперативном лечении слоновоподобной формы нейрофиброматоза требуется последующая кожная пластика. Ткань нейрофибром обильно снабжена кровеносными сосудами. При расположении узла в крупном нервном стволе производят вылущивание опухоли, резекцию нерва с наложением нервного шва или краевую его резекцию с наложением частичного нервного шва. Оперативное удаление одного из узлов в ряде случаев может привести к прогрессированию процесса с резким увеличением размеров других узлов[36].
Патогенетическое лечение (то есть направленное на основные механизмы развития заболевания) на первую половину 2011 года находится на I—II фазах клинических исследований[22] и повсеместно не применяется. Имеются данные об эффективности ингибиторов Ras (типифарниба) в лечении нейрофиброматоза I типа[45]. Также на животных показана эффективность пирфенидона[46]. Однако до завершения клинических исследований эти и ряд других препаратов не могут использоваться в лечении нейрофиброматоза.
В апреле 2020 года FDA одобрило «Коселуго» (Koselugo, селуметиниб) — первый лекарственный препарат, предназначенный для терапии детей в возрасте от двух лет, страдающих нейрофиброматозом I типа. Заболевание пациентов должно быть симптоматическим и характеризоваться наличием неоперабельных плексиформных нейрофибром. Селуметиниб (selumetinib), разработанный «Эрей байофарма» (Array BioPharma), которая лицензировала его «АстраЗенека» (AstraZeneca), представляет собой пероральный низкомолекулярный АТФ-независимый ингибитор киназы митоген-активируемой протеинкиназы типа 1 (MEK1) и типа 2 (MEK2). Белки MEK1 и MEK2, необходимые для активации сигнального пути RAS/RAF/MEK/ERK, зачастую проявляют повышенную активность, которая отражается в том числе клеточной пролиферацией. Подавление MEK1/MEK2 приводит к ингибированию ERK-фосфорилирования, что результирует снижением числа нейрофибром и уменьшением их объёмов, сдерживанием пролиферации клеток[47].
Прогноз
Ожидаемая продолжительность жизни снижена на 8—12 лет в сравнении с общей популяцией. Повышение смертности происходит у более молодых лиц (в возрасте до 40 лет), преимущественно за счёт повышенного риска появления злокачественных новообразований[48].
См. также
- Нейрофиброматоз
- Костная болезнь Реклингхаузена
Примечания
- ↑ 1 2 3 4 5 6 7 8 9 Козлов А. В. Нейрофиброматоз 1 (НФ1) // Хирургия опухолей основания черепа / Под редакцией А. Н. Коновалова. — М.:: ОАО «Можайский полиграфический комбинат», 2004. — С. 166—169. — 372 с. — 1000 экз. — ISBN 5-94982-006-1.
- ↑ Нейрофиброматоз на neuro-med.ru. Дата обращения: 16 августа 2010. Архивировано 1 февраля 2012 года.
- ↑ NEUROFIBROMATOSIS, TYPE I; NF1 — OMIM Result. Дата обращения: 3 октября 2017. Архивировано 15 апреля 2011 года.
- ↑ Шнайдер Н. А. Нейрофиброматоз. сайт krasmedic.ru. Дата обращения: 15 мая 2011. Архивировано 26 февраля 2021 года.
- ↑ Нейрофиброматоз – виды заболевания. сайт doctoram.net. Дата обращения: 15 мая 2011. Архивировано из оригинала 17 августа 2011 года.
- ↑ Нейрофиброматоз (болезнь Реклингхаузена). сайт myemergency.ru. Дата обращения: 13 мая 2011. Архивировано 18 ноября 2012 года.
- ↑ Нейрофиброматоз (болезнь Реклингхаузена). сайт www.medfix.ru. Дата обращения: 13 мая 2011. Архивировано 2 марта 2015 года.
- ↑ Ahn MS, Jackler RK, Lustig LR. The early history of the neurofibromatosis. Evolution of the concept of neurofibromatosis type 2. // Arch Otolaryngol Head Neck Surg. — 1996. — Т. 122. — С. 1240—1249. — PMID 8906061.
- ↑ Elephant man mystery unravelled (англ.). BBC News (21 июля 2003). Дата обращения: 13 мая 2011. Архивировано 17 августа 2011 года.
- ↑ Roger Highfield. Science uncovers handsome side of the Elephant Man (англ.). The Telegraph (13 мая 2003). Дата обращения: 13 мая 2011. Архивировано 17 августа 2011 года.
- ↑ 1 2 Шнайдер Н. А. ДНК-диагностика нейрофиброматоза 1 типа (болезни Реклингхаузена). сайт www.krasmedic.ru. Дата обращения: 6 мая 2011. Архивировано 1 марта 2021 года.
- ↑ M. Deckert, G. Reifenberger, U.-N. Riede, W. Schlote, D.R. Thal, O.D. Wiestler. Nervensystem // Allgemeine und Spezielle Pathologie / Ursus-Nikolaus Riede, Martin Werner, Hans-Eckart Schäfer. — 5. — Stuttgart, 2004. — P. 1104.
- ↑ Ponder B. Human genetics. Neurofibromatosis gene cloned // Nature. — 1990. — Т. 346. — С. 703—704. — PMID 2117711. Архивировано 6 июня 2011 года.
- ↑ Viskochil D, Buchberg AM, Xu G, Cawthon RM, Stevens J, Wolff RK, Culver M, Carey JC, Copeland NG, Jenkins NA, et al. Deletions and a translocation interrupt a cloned gene at the neurofibromatosis type 1 locus. (англ.) // Cell. — 1990. — Vol. 62. — P. 187—192. — PMID 1694727. Архивировано 20 мая 2016 года.
- ↑ Нейрофиброматоз. сайт «База знаний по биологии человека». Дата обращения: 7 мая 2011. Архивировано из оригинала 7 октября 2010 года.
- ↑ Wallace MR, Marchuk DA, Andersen LB, Letcher R, Odeh HM, Saulino AM, Fountain JW, Brereton A, Nicholson J, Mitchell AL, et al. Type 1 neurofibromatosis gene: identification of a large transcript disrupted in three NF1 patients. (англ.) // Science. — 1990. — Vol. 249. — P. 181—186. — PMID 2134734. Архивировано 28 мая 2016 года.
- ↑ Abernathy CR, Rasmussen SA, Stalker HJ, Zori R, Driscoll DJ, Williams CA, Kousseff BG, Wallace MR. NF1 mutation analysis using a combined heteroduplex/SSCP approach. // Hum Mutat.. — 1997. — Т. 9. — С. 548—54. — PMID 9195229.
- ↑ Kluwe L., Tatgiba M., Fünsterer C., Mautner V.-F. [www.ncbi.nlm.nih.gov/pmc/articles/PMC1735482/pdf/v040p00368.pdf NF1 mutations and clinical spectrum in patients with spinal neurofibromas] // J Med Genet. — 2003. — Т. 40. — С. 368—371.
- ↑ Osborn MJ, Upadhyaya M. Evaluation of the protein truncation test and mutation detection in the NF1 gene: mutational analysis of 15 known and 40 unknown mutations // Hum Genet.. — 1999. — Т. 105. — С. 327—332. — PMID 10543400.
- ↑ Ars E, Serra E, de la Luna S, Estivill X, Lázaro C. Cold shock induces the insertion of a cryptic exon in the neurofibromatosis type 1 (NF1) mRNA // Nucleic Acids Res.. — 2000. — Т. 28. — С. 1307—1312. — PMID 10684924.
- ↑ Messiaen LM, Callens T, Mortier G, Beysen D, Vandenbroucke I, Van Roy N, Speleman F, Paepe AD. Exhaustive mutation analysis of the NF1 gene allows identification of 95% of mutations and reveals a high frequency of unusual splicing defects // Hum Mutat.. — 2000. — Т. 15. — С. 541—555. — PMID 10862084.
- ↑ 1 2 3 4 5 Williams V.C., Lucas J., Babcock M.A. et al. Neurofibromatosis Type 1 Revisited (англ.) // Pediatrics. — 2009. — Vol. 123. — P. 124—133. Архивировано 4 марта 2016 года.
- ↑ Xu GF, O'Connell P, Viskochil D, Cawthon R, Robertson M, Culver M, Dunn D, Stevens J, Gesteland R, White R, et al. The neurofibromatosis type 1 gene encodes a protein related to GAP (англ.) // Cell. — 1990. — Vol. 62. — P. 599—608. — PMID 2116237. Архивировано 3 июня 2016 года.
- ↑ 1 2 3 4 5 6 7 8 9 10 11 12 Шнайдер Н. А. Нейрофиброматоз 1-го типа: этиопатогенез, клиника, диагностика, прогноз // Международный неврологический журнал. — 2007. — № 5 (15). Архивировано 18 апреля 2011 года.
- ↑ Doran S.E., Thorell W.E. Chapter 40. Brain Tumors // Youmans Neurological surgery / Winn H.R.. — 5th edition. — Philadelphia: Saunders, 2004. — Vol. 1. — P. 812. — ISBN 0-7216-8291-x.
- ↑ Соловьёв Ю. Н. Нейрофиброма // Большая медицинская энциклопедия / под общей редакцией Б. В. Петровского. — 3-е издание. — М.:: «Советская энциклопедия», 1981. — Т. 16. — С. 309—310. — 512 с. — 150 000 экз.
- ↑ Riccardi VM. Von Recklinghausen neurofibromatosis (англ.) // N Engl J Med.. — 1981. — Vol. 305. — P. 1617—1627. — PMID 6796886.
- ↑ Dutton J. J. Gliomas of the anterior visual pathway (англ.) // Surv Ophtalmol. — 1994. — Vol. 38. — P. 427—452. — PMID 8009427.
- ↑ Jahraus C. D., Tarbell N. J. Optic pathway gliomas (англ.) // Pediatr Blood Cancer. — 2006. — Vol. 46. — P. 586—596. — PMID 16411210.
- ↑ 1 2 Özek M. M., Urgun K. Tumors of the Optic Pathway and Hypothalamus // Essential Practice of Neurosurgery / Editor-in-Chief Kazadi K. N. Kalangu, Deputy Editors Yoko Kato and Gilbert Dechambenoit. — Nagoya, Japan: Yamagiku Printing Co., Ltd. — P. 348. — 1496 p. — ISBN 978-4-904992-01-2.
- ↑ Davis F. A. Primary tumours of the optic nerve (a phenomenon of Recklinghausen`s disease) (англ.) // Arch Ophth. — 1940. — Vol. 23. — P. 957—1018.
- ↑ Bondy M., Wrensch M., Wiencke J. Genetic and hereditary syndromes associated with tumors of the CNS // Cancer in the Nervous System / Levin. — 5th edition. — New York: Churchill Livingstone, 1996. — P. 1—12.
- ↑ Roach ES. Diagnosis and management of neurocutaneous syndromes // Semin Neurol.. — 1988. — Т. 8. — С. 83—96. — PMID 3146120.
- ↑ Sørensen SA, Mulvihill JJ, Nielsen A. Long-term follow-up of von Recklinghausen neurofibromatosis. Survival and malignant neoplasms // N Engl J Med.. — 1986. — Т. 314. — С. 1010—1015. — PMID 3083258.
- ↑ Blatt J, Jaffe R, Deutsch M, Adkins JC. Neurofibromatosis and childhood tumors // Cancer. — 1986. — Т. 57. — С. 1225—1229. — PMID 3080222.
- ↑ 1 2 3 4 5 6 Мельников Р. А., Китаев В. В. Нейрофиброматоз // Большая медицинская энциклопедия / под общей редакцией Б. В. Петровского. — 3-е издание. — М.:: "Советская энциклопедия", 1981. — Т. 16. — С. 310—311. — 512 с. — 150 000 экз.
- ↑ 1 2 Нейрофиброматоз Реклингхаузена. сайт lechenee.ru. Дата обращения: 7 мая 2011. (недоступная ссылка)
- ↑ P. J. Waardenburg. Heterochrome en melanosis // Nederlands tijdschrift voor geneeskunde. — Haarlem, 1918. — Т. 62. — С. 1453—1455.
- ↑ Riccardi VM. Neurofibromatosis: an overview and new directions in clinical investigations // Adv Neurol.. — 1981. — Т. 29. — С. 1—9. — PMID 6798831.
- ↑ Lubs ML, Bauer MS, Formas ME, Djokic B. Lisch nodules in neurofibromatosis type 1 // N Engl J Med.. — 1991. — Т. 324. — С. 1264—1266. — PMID 1901624.
- ↑ Симптомы и синдромы в эндокринологии / Под ред. Ю. И. Караченцева. — 1-е изд. — Х.: ООО «С.А.М.», Харьков, 2006. — С. 141. — 227 с. — (Справочное пособие). — 1000 экз. — ISBN 978-966-8591-14-3.
- ↑ Neurofibromatosis. Conference Statement, NIH Consensus Development Conference. in: Archives of neurology. official organ of the American Neurological Association. Chicago Ill 45.1988, 575—578
- ↑ Нейрофиброматоз. сайт emed.nextday.su. Дата обращения: 7 мая 2011. (недоступная ссылка)
- ↑ Козлов А. В. Нейрофиброматоз 1 (НФ1) // Хирургия опухолей основания черепа / Под редакцией А. Н. Коновалова. — М.:: ОАО «Можайский полиграфический комбинат», 2004. — С. 169—170. — 372 с. — 1000 экз. — ISBN 5-94982-006-1.
- ↑ Widemann BC, Salzer WL, Arceci RJ, Blaney SM, Fox E, End D, Gillespie A, Whitcomb P, Palumbo JS, Pitney A, Jayaprakash N, Zannikos P, Balis FM. Phase I trial and pharmacokinetic study of the farnesyltransferase inhibitor tipifarnib in children with refractory solid tumors or neurofibromatosis type I and plexiform neurofibromas (англ.) // J Clin Oncol. — 2006. — Vol. 24. — P. 507—516. — PMID 16421428.
- ↑ Babovic-Vuksanovic D, Petrovic L, Knudsen BE, Plummer TB, Parisi JE, Babovic S, Platt JL. Survival of Human Neurofibroma in Immunodeficient Mice and Initial Results of Therapy With Pirfenidone (англ.) // J Biomed Biotechnol.. — 2004. — Vol. 2. — P. 79—85. — PMID 15240917.
- ↑ Татьяна Фон Ройсс. «Коселуго»: первое лекарство против нейрофиброматоза. Селуметиниб мощно подавляет рост опухолей. Mosmedpreparaty.ru. Мосмедпрепараты (9 июля 2020). Дата обращения: 18 ноября 2020. Архивировано 30 сентября 2020 года.
- ↑ David H. Gutmann, Rosalie E. Ferner, Robert H. Listernick, Bruce R. Korf, Pamela L. Wolters. Neurofibromatosis type 1 (англ.) // Nature Reviews Disease Primers. — 2017-02-23. — Vol. 3, iss. 1. — P. 1–17. — ISSN 2056-676X. — doi:10.1038/nrdp.2017.4. Архивировано 17 октября 2022 года.
Синдромы в эндокринологии | |
|---|---|
| Эпифиз |
|
| Гипоталамус |
|
| Гипофиз |
|
| Щитовидная железа |
|
| Надпочечники |
|
| Половые железы |
|
| Паращитовидные железы | |
| Островки Лангерганса | |
| Диффузная нейроэндокринная система |
|
| Прочие |
|
| |||||||||||||||||||||||
| |||||||||||||||||||||||
| |||||||||||||||||||||||