Одномолекулярное секвенирование в реальном времени
Одномолекуля́рное секвени́рование в реа́льном вре́мени (англ. Single molecule real time sequencing или SMRT) — метод секвенирования ДНК нового поколения, разработанный компанией Pacific Biosciences[англ.].
Идея метода состоит в определении последовательности ДНК за счёт наблюдения за работой единичной молекулы ДНК-полимеразы в реальном времени. При этом ДНК-полимераза достраивает вторую цепь исследуемой молекулы ДНК, используя нуклеотиды, меченные различными флуоресцентными метками[англ.]; регистрируя данные метки, можно понять, какой нуклеотид ДНК-полимераза встраивает в настоящий момент.
Технология
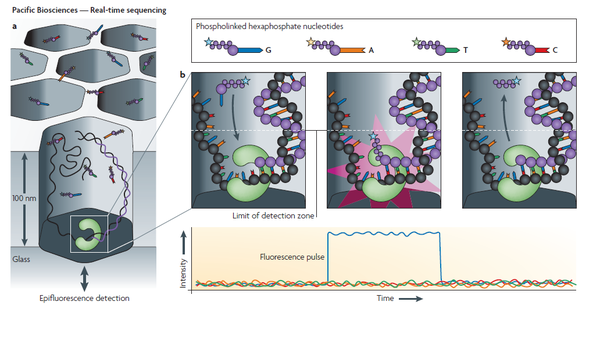
Устройство секвенаторов данного типа позволяет наблюдать на уровне единичной молекулы за синтезом комплементарной цепи одной молекулы одноцепочечной ДНК с помощью одной молекулы ДНК-полимеразы. В этой технологии флуоресцентно меченные нуклеотиды и конфокальная микроскопия высокого разрешения позволяют секвенировать последовательность в реальном времени и одновременно для многих полимераз[1].
ZMW
В основе метода лежит использование Zero-mode waveguide (ZMW) — углублений диаметром несколько десятков нанометров, ко дну которых прикреплена единичная молекула ДНК-полимеразы. Сквозь дно в ZMW-ячейку подаётся свет. Особенность конструкции ZMW-ячейки не даёт распространяться световой волне и оставляет освещённым только объём порядка 20 цептолитров (20 × 10−21 литров) около дна ячейки. Это позволяет наблюдать флуоресценцию единичной флуоресцентной метки, пришитой к нуклеотиду, в данный момент встраиваемому ДНК-полимеразой. Соответственно к четырем типам нуклеотидов пришиты разные флуоресцентные метки, что позволяет различать их. В результате при полимеризации цепи ДНК зафиксированным в ZMW ферментом можно получить зависимость интенсивности флуоресценции от времени, из графика которой по пикам разного спектра определяется последовательность ДНК[1].
При секвенировании используются так называемые SMRT cells, содержащие порядка 150000 ZMW-ячеек, которые представляют собой углубления в алюминиевой плёнке, напылённой на кремниевую подложку[2].
Нуклеотиды
В данном методе используют метки (флуорофоры), пришитые к терминальной фосфатной группе нуклеотид. Такая метка меньше влияет на работу ДНК-полимеразы, что крайне важно при секвенировании в реальном времени. В процессе присоединения нуклеотида к растущей цепи ДНК метка отщепляется ДНК-полимеразой вместе с пирофосфатом. В результате этого флуорофор может диффундировать из наблюдаемого объёма и больше не влиять на регистрируемый сигнал, а нуклеотид без «довесков» встраивается в цепь ДНК. Таким образом, измеряя длительное (миллисекунды) свечение одного цвета при присоединении полимеразой меченного нуклеотида на фоне быстро диффундирующих (микросекунды) четырёх, можно определить последовательность матричной цепи ДНК[1].
Преимущества метода
Метод одномолекулярного секвенирования в реальном времени даёт возможность получать очень длинные чтения (последовательности ДНК) (в среднем порядка 20000 нуклеотидов, вплоть до 60000 нуклеотидов), что облегчает дальнейший анализ данных и позволяет избежать ряд проблем, возникающих при работе с короткими чтениями. Он работает без предварительной амплификации исследуемой ДНК посредством ПЦР. Этот метод обеспечивает высокую скорость секвенирования (в теории она ограничена только скоростью работы ДНК-полимеразы)[1]. Для метода характерны высокая чувствительность и специфичность: возможность детектирования в смешанных образцах минорных вариантов с частотой встречаемости меньше 0,1 %. Он также даёт возможность секвенирования с высокой точностью. На данный момент она не очень высока (83 %), но точность можно повысить за счёт повторного секвенирования молекулы ДНК (> 99 % при 15 повторениях)[3][4].
Недостатки метода
К недостаткам метода можно отнести высокую стоимость прибора — 600000 $[5]. Для него характерен сравнительно высокий уровень ошибок, обусловленный пересечением спектров излучения флуорофоров. Кроме того, случайное присоединение полимераз ко дну ZMW-ячейки приводит к пуассоновскому распределению числа ферментов на одну ячейку[1].
Применение
Секвенирование de novo
Длина чтений одномолекулярного секвенирования в реальном времени сравнима или больше, чем в методе Сэнгера, что даёт возможность секвенировать геномы de novo[англ.] и упрощает их сборку[1]. Длинные чтения обеспечивают контекст, необходимый для правильного определения позиций повторов в геноме. Возможность получения длинных участков ДНК при секвенировании важна также для метагеномики: можно идентифицировать организмы в смешанных популяциях — например, в микробиоме. Поскольку для сборки генома необходимо меньшее количество прочтений одних и тех же участков, расшифровка генома таким методом требует меньших затрат. Одномолекулярное секвенирование в реальном времени было продемонстрировано на секвенировании геномов de novo в работах, посвящённых анализу вспышки кишечной инфекции в Германии в 2011 году и эпидемии холеры на Гаити в 2010 году[6][7].
Гибридная корректировка сборки геномов
Технология секвенирования «третьего поколения» в сочетании с более старыми методами позволяет увеличить точность сборки генома. Секвенаторы «второго поколения» способны считывать геном небольшими фрагментами протяженностью 100—700 пар оснований, но такие чтения затем сложно соединить в правильном порядке. Приборы «третьего поколения» (в частности, PacBio RS компании Pacific Biosciences) могут генерировать чтения длиной до 23 тыс. пар оснований, но делают больше ошибок, чем допустимо для обычного программного обеспечения для геномного анализа. В 2011 году учёные из центра National Biodefense Analysis and Countermeasures Center[англ.] (США) использовали короткие чтения, полученные в ходе секвенирования на приборах второго поколения Illumina и Roche 454, для корректировки ошибок в длинных чтениях, генерированных секвенатором PacBio RS. Протестировав разработанный алгоритм на геномах бактерии Escherichia coli и дрожжей, а также на транскриптоме кукурузы, исследователи выяснили, что точность сборки можно повысить с 83 до 99,9 %. Ученые также применили разработанный метод гибридной корректировки к сборке ранее несеквенированного генома волнистого попугайчика[8].
В 2012 году гибридный подход был использован для сборки генома штамма холеры, вызвавшего эпидемию на Гаити в 2010 году. Участки генома бактерии, важные с точки зрения лечения заболевания, были собраны с точностью, превосходящей 99,9 %[9].
Ресеквенирование и определение геномных вариаций
Одна и та же молекула ДНК может быть независимо ресеквенирована с помощью кольцевой ДНК-матрицы и фермента, отделяющего вновь синтезированную цепь ДНК от матрицы. Это имеет важное значение для анализа и диагностики различных заболеваний. Сравнивая миллионы и миллиарды чтений с исходным текстом, можно получить полный перечень отличий изучаемого генома от «золотого стандарта». Более того, если каждая буква исходного текста проверяется многократными прочтениями, это увеличивает статистическую достоверность найденных генетических особенностей и аномалий[10].
Учёные компании Pacific Biosciences совместно со специалистами из других организаций использовали данный подход для обоснования гипотезы об активирующей тандемной дупликации FLT3 как терапевтической мишени при остром миелолейкозе[10]. Данная технология также подходит для анализа транскриптома и сплайсинга, поскольку одно длинное чтение с секвенатора может содержать целую мРНК. Одномолекулярное секвенирование в реальном времени с высокой точностью позволяет детектировать однонуклеотидные полиморфизмы[11].
Расшифровка эпигенома
Кинетика реакции полимеризации в ходе секвенирования даёт возможность определять ключевые эпигенетические модификации ДНК. При одномолекулярном секвенировании в реальном времени о наличии метилированных нуклеотидов судят по изменению периода до следующей вспышки, так как метилирование влияет на активность полимеразы. Этот метод уже используется для определения метиладенина[англ.], метилцитозина[англ.], а также 5-гидроксиметилцитозина[англ.][12][13][14]. В 2012 году группа ученых использовала данный подход для анализа полного профиля метилирования 6 бактерий[15].
Секвенирование РНК
Используя вместо ДНК-полимеразы обратную транскриптазу, SMRT технология позволяет секвенировать РНК. Таким способом можно одновременно детектировать последовательность, модификации оснований, перестановки, влияющие на структуру РНК. Кинетика обратной транскрипции чувствительна также к вторичной структуре РНК, которая увеличивает вероятность долгих пауз или терминации в ходе реакции. Более того, SMRT секвенирование позволяет детектировать динамику рефолдинга РНК — например, в процессе обратной транскрипции ретровирусов или во время деградации мРНК экзосомами[16].
Коммерческое использование
Компания Pacific Biosciences|Pacific Biosciences коммерциализировала SMRT-секвенирование в 2011 году[17] после выпуска второго варианта установки в конце 2010 года[18].
В апреле 2013 года компания выпустила новую версию секвенатора под названием «PacBio RS II», который имеет более высокую пропускную способность и даёт возможность получать более длинные чтения ДНК[19][20].
Прототип чипа SMRT содержал ~ 3000 ZMW-ячеек для параллельного секвенирования ДНК. В 2012 году были созданы SMRT cells, каждый из которых содержал около 150000 ZMW-ячеек[21].
Новый набор реактивов, выпущенный в 2012 году, позволил увеличить длину чтения[22]. На данный момент средняя длина прочтения составляет около 40 000 н. п., максимальная — 100 000 н. п[23].
Примечания
- ↑ 1 2 3 4 5 6 Eid J., Fehr A., Gray J., Luong K., Lyle J., Otto G., Peluso P., Rank D., Baybayan P., Bettman B., Bibillo A., Bjornson K., Chaudhuri B., Christians F., Cicero R., Clark S., Dalal R., Dewinter A., Dixon J., Foquet M., Gaertner A., Hardenbol P., Heiner C., Hester K., Holden D., Kearns G., Kong X., Kuse R., Lacroix Y., Lin S., Lundquist P., Ma C., Marks P., Maxham M., Murphy D., Park I., Pham T., Phillips M., Roy J., Sebra R., Shen G., Sorenson J., Tomaney A., Travers K., Trulson M., Vieceli J., Wegener J., Wu D., Yang A., Zaccarin D., Zhao P., Zhong F., Korlach J., Turner S. Real-time DNA sequencing from single polymerase molecules. (англ.) // Science (New York, N.Y.). — 2009. — Vol. 323, no. 5910. — P. 133—138. — doi:10.1126/science.1162986. — PMID 19023044.
- ↑ SMRT-cells. Дата обращения: 2 мая 2013. Архивировано 21 апреля 2013 года.
- ↑ Metzker M. L. Sequencing technologies - the next generation. (англ.) // Nature reviews. Genetics. — 2010. — Vol. 11, no. 1. — P. 31—46. — doi:10.1038/nrg2626. — PMID 19997069.
- ↑ Pacific Biosciences: SMRT Sequencing Advantage. Дата обращения: 9 мая 2013. Архивировано из оригинала 19 мая 2013 года.
- ↑ Quail M. A., Smith M., Coupland P., Otto T. D., Harris S. R., Connor T. R., Bertoni A., Swerdlow H. P., Gu Y. A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. (англ.) // BMC genomics. — 2012. — Vol. 13. — P. 341. — doi:10.1186/1471-2164-13-341. — PMID 22827831.
- ↑ Chin C. S., Sorenson J., Harris J. B., Robins W. P., Charles R. C., Jean-Charles R. R., Bullard J., Webster D. R., Kasarskis A., Peluso P., Paxinos E. E., Yamaichi Y., Calderwood S. B., Mekalanos J. J., Schadt E. E., Waldor M. K. The origin of the Haitian cholera outbreak strain. (англ.) // The New England journal of medicine. — 2011. — Vol. 364, no. 1. — P. 33—42. — doi:10.1056/NEJMoa1012928. — PMID 21142692.
- ↑ Rasko D. A., Webster D. R., Sahl J. W., Bashir A., Boisen N., Scheutz F., Paxinos E. E., Sebra R., Chin C. S., Iliopoulos D., Klammer A., Peluso P., Lee L., Kislyuk A. O., Bullard J., Kasarskis A., Wang S., Eid J., Rank D., Redman J. C., Steyert S. R., Frimodt-Møller J., Struve C., Petersen A. M., Krogfelt K. A., Nataro J. P., Schadt E. E., Waldor M. K. Origins of the E. coli strain causing an outbreak of hemolytic-uremic syndrome in Germany. (англ.) // The New England journal of medicine. — 2011. — Vol. 365, no. 8. — P. 709—717. — doi:10.1056/NEJMoa1106920. — PMID 21793740.
- ↑ Koren S., Schatz M. C., Walenz B. P., Martin J., Howard J. T., Ganapathy G., Wang Z., Rasko D. A., McCombie W. R., Jarvis E. D., Adam M. Phillippy. Hybrid error correction and de novo assembly of single-molecule sequencing reads. (англ.) // Nature biotechnology. — 2012. — Vol. 30, no. 7. — P. 693—700. — doi:10.1038/nbt.2280. — PMID 22750884.
- ↑ Bashir A., Klammer A. A., Robins W. P., Chin C. S., Webster D., Paxinos E., Hsu D., Ashby M., Wang S., Peluso P., Sebra R., Sorenson J., Bullard J., Yen J., Valdovino M., Mollova E., Luong K., Lin S., LaMay B., Joshi A., Rowe L., Frace M., Tarr C. L., Turnsek M., Davis B. M., Kasarskis A., Mekalanos J. J., Waldor M. K., Schadt E. E. A hybrid approach for the automated finishing of bacterial genomes. (англ.) // Nature biotechnology. — 2012. — Vol. 30, no. 7. — P. 701—707. — doi:10.1038/nbt.2288. — PMID 22750883.
- ↑ 1 2 Smith C. C., Wang Q., Chin C. S., Salerno S., Damon L. E., Levis M. J., Perl A. E., Travers K. J., Wang S., Hunt J. P., Zarrinkar P. P., Schadt E. E., Kasarskis A., Kuriyan J., Shah N. P. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. (англ.) // Nature. — 2012. — Vol. 485, no. 7397. — P. 260—263. — doi:10.1038/nature11016. — PMID 22504184.
- ↑ Carneiro M. O., Russ C., Ross M. G., Gabriel S. B., Nusbaum C., DePristo M. A. Pacific biosciences sequencing technology for genotyping and variation discovery in human data. (англ.) // BMC genomics. — 2012. — Vol. 13. — P. 375. — doi:10.1186/1471-2164-13-375. — PMID 22863213.
- ↑ Clark T. A., Murray I. A., Morgan R. D., Kislyuk A. O., Spittle K. E., Boitano M., Fomenkov A., Roberts R. J., Korlach J. Characterization of DNA methyltransferase specificities using single-molecule, real-time DNA sequencing. (англ.) // Nucleic acids research. — 2012. — Vol. 40, no. 4. — P. e29. — doi:10.1093/nar/gkr1146. — PMID 22156058.
- ↑ Song C. X., Clark T. A., Lu X. Y., Kislyuk A., Dai Q., Turner S. W., He C., Korlach J. Sensitive and specific single-molecule sequencing of 5-hydroxymethylcytosine. (англ.) // Nature methods. — 2011. — Vol. 9, no. 1. — P. 75—77. — doi:10.1038/nmeth.1779. — PMID 22101853.
- ↑ Clark T. A., Spittle K. E., Turner S. W., Korlach J. Direct detection and sequencing of damaged DNA bases. (англ.) // Genome integrity. — 2011. — Vol. 2. — P. 10. — doi:10.1186/2041-9414-2-10. — PMID 22185597.
- ↑ Murray I. A., Clark T. A., Morgan R. D., Boitano M., Anton B. P., Luong K., Fomenkov A., Turner S. W., Korlach J., Roberts R. J. The methylomes of six bacteria. (англ.) // Nucleic acids research. — 2012. — Vol. 40, no. 22. — P. 11450—11462. — doi:10.1093/nar/gks891. — PMID 23034806.
- ↑ Vilfan I. D., Tsai Y. C., Clark T. A., Wegener J., Dai Q., Yi C., Pan T., Turner S. W., Korlach J. Analysis of RNA base modification and structural rearrangement by single-molecule real-time detection of reverse transcription. (англ.) // Journal of nanobiotechnology. — 2013. — Vol. 11. — P. 8. — doi:10.1186/1477-3155-11-8. — PMID 23552456.
- ↑ PacBio Ships First Two Commercial Systems; Order Backlog Grows to 44. Дата обращения: 9 мая 2013. Архивировано 27 сентября 2013 года.
- ↑ PacBio Reveals Beta System Specs for RS; Says Commercial Release is on Track for First Half of 2011. Дата обращения: 9 мая 2013. Архивировано 27 сентября 2013 года.
- ↑ PacBio Launches PacBio RS II Sequencer. Дата обращения: 9 мая 2013. Архивировано 19 декабря 2019 года.
- ↑ New Products: PacBio's RS II; Cufflinks. Дата обращения: 9 мая 2013. Архивировано 2 мая 2013 года.
- ↑ Pacific Biosciences: Consumables. Дата обращения: 2 мая 2013. Архивировано 21 апреля 2013 года.
- ↑ After a Year of Testing, Two Early PacBio Customers Expect More Routine Use of RS Sequencer in 2012. Дата обращения: 9 мая 2013. Архивировано 12 декабря 2013 года.
- ↑ Официальный сайт Pacific Biosciences. Дата обращения: 1 мая 2013. Архивировано 3 мая 2013 года.
См. также
Ссылки
- Сайт компании Pacific Biosciences. Дата обращения: 1 мая 2013. Архивировано 3 мая 2013 года.
- Рекламная анимация компании Pacific Biosciences, описывающая суть метода.