
Боле́знь Паркинсо́на — медленно прогрессирующее хроническое нейродегенеративное неврологическое заболевание, характерное для лиц старшей возрастной группы. Относится к дегенеративным заболеваниям экстрапирамидной моторной системы. Вызвано прогрессирующим разрушением и гибелью нейронов, вырабатывающих нейромедиатор дофамин, — прежде всего в чёрной субстанции, а также и в других отделах центральной нервной системы. Недостаточная выработка дофамина ведёт к тормозному влиянию базальных ганглиев на кору головного мозга. Ведущими симптомами являются:
- мышечная ригидность;
- гипокинезия;
- тремор;
- постуральная неустойчивость.

Целиаки́я, глютеновая энтеропатия — форма энтеропатии, генетически предрасположенная непереносимость глиадина или глютена. Это аутоиммунное заболевание с преимущественной локализацией в тонком кишечнике.
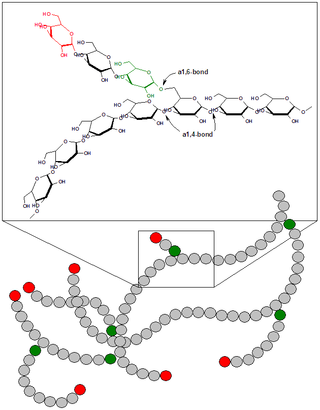
Боле́знь Герса — гепатофосфорилазная недостаточность, гликогеноз, вызванный недостаточностью фосфорилазы печени.

Болезнь Гоше́ — наследственное заболевание типа сфинголипидозов, является самой распространённой из лизосомных болезней накопления. Развивается в результате недостаточности фермента глюкоцереброзидазы, которая приводит к накоплению глюкоцереброзида во многих тканях, включая селезёнку, печень, почки, лёгкие, мозг и костный мозг. Заболевание связано с рецессивной мутацией в гене GBA, расположенном в 1-й хромосоме, и поражает как мужчин, так и женщин. Заболевание названо в честь французского врача Филиппа Гоше, который первым описал его в 1882.
Орфа́нные препара́ты — фармацевтические средства, разработанные для лечения редких заболеваний, которые условно называются орфанными («сиротскими») болезнями, как правило, коммерчески нерентабельные для фармацевтической индустрии и научно-исследовательских лабораторных подразделений фармацевтических компаний, а потому разрабатываемые в последнюю очередь.
Болезнь Краббе — редкое наследственное заболевание, при котором поражается миелиновая оболочка нервных волокон. Наследуется аутосомно-рецессивно. Болезнь названа в честь датского невролога Краббе, который описал её в 1916 году.

Синдром Шарпа, или смешанное заболевание соединительной ткани , — аутоиммунное заболевание, при котором защитная система организма атакует саму себя. Впервые синдром описан в 1972 году.
Паренхимато́зные дистрофи́и — нарушения метаболизма в паренхиме органов.
Помпе:
- Помпе — коммуна во французском департаменте Мёрт и Мозель, региона Лотарингия.
- Помпе — кантон во Франции, находится в регионе Лотарингия.
- Помпе, Иоанн Кассианус (1901—1945) — голландский патолог.
- Болезнь Помпе — редкое наследственное заболевание, описанное Иоанном Кассианусом Помпе.
Нейродегенеративные заболевания — группа в основном медленно прогрессирующих, наследственных или приобретённых заболеваний нервной системы. Общим для этих заболеваний является прогрессирующая гибель нервных клеток (нейродегенерация), ведущая к различным неврологическим симптомам — прежде всего, к деменции и нарушению движений. Заболевания могут наступить в различном возрасте, протекают диффузно или генерализированно, гистологически определяется специфический тип изменений.

Лизосо́мные боле́зни накопле́ния — общее название группы весьма редких наследственных заболеваний, вызванных нарушением функции внутриклеточных органелл лизосом. Эти одномембранные органоиды являются частью эндомембранной системы клетки и специализируются на внутриклеточном расщеплении веществ: гликогена, гликозаминогликанов, гликопротеинов и других. Лизосомные болезни накопления вызываются генетически обусловленным дефицитом ферментов лизосом, что приводит к накоплению макромолекул, являющихся субстратом этих ферментов, в различных органах и тканях организма.

Сфинголипидо́зы группа лизосомных болезней накопления, связанных с нарушением метаболизма сфинголипидов, относится к классу болезней накопления липидов (липидозов). Основными представителями этой группы являются болезнь Ниманна — Пика, болезнь Фабри, болезнь Краббе, болезнь Гоше, болезнь Тея — Сакса и метахроматическая лейкодистрофия. Они, как правило, наследуется по аутосомно-рецессивному типу, однако в частности, болезнь Фабри — редкое генетически детерминированное заболевание с Х-сцепленным рецессивным типом наследования.

Боле́знь По́мпе — редкое, наследственное заболевание с аутосомно-рецессивным механизмом, наследования,, связанное с повреждением мышечных, и нервных клеток по всему организму. Клиническая картина данной патологии, обусловлена накоплением гликогена, в лизосомах, вызванным недостаточностью лизосомного фермента — кислой α-1,4-глюкозидазы. Различают быстро прогрессирующую (классическую) и медленно прогрессирующую формы болезни Помпе. Несмотря на широкий спектр клинических проявлений, гликогеноза II типа, в основе всех форм болезни лежит, дефицит одного фермента, кодируемого геном GAA.

Боле́знь Фабри́ — редкое генетически детерминированное заболевание с Х-сцепленным типом наследования, из группы лизосомных болезней накопления. Ранее считалось, что тип наследования болезни Фабри — X-сцепленный рецессивный, однако на современном этапе накоплено достаточно данных, чтобы считать тип наследования болезни Фабри X-сцепленным доминантным с неполной пенетрантностью у женщин. Данное заболевание вызвано нарушением метаболизма сфинголипидов и обладает широким спектром клинических симптомов.

Воскови́дные липофусцино́зы нейро́нов — общее название широкой группы нейродегенеративных наследственных заболеваний, относящихся к лизосомным болезням накопления. Симптомы болезней данной группы обусловлены чрезмерным накоплением пигмента липофусцина в лизосомах нервных клеток и многих других тканей организма, включая печень, селезёнку, миокард, почки. Избыточное отложение липофусцина в лизосомах вызывает зеленовато-жёлтое окрашивание при микроскопии в ультрафиолетовых лучах.
GM2 ганглиозидо́з, вариа́нт АБ (англ. GM2-gangliosidosis, AB variant) — редкое аутосомно-рецессивное нарушение обмена веществ, связанное с мутацией гена GM2A. Характеризуется нормальной активностью β-гексозаминидаз А и Б и обусловлено недостаточностью активатора (белкового кофактора), необходимого для реализации ферментной активности в отношении субстрата. Заболевание клинически проявляется прогрессирующим разрушением нервных клеток головного и спинного мозга.
Болезнь Сантавуори — Халтиа — редкое наследственное нейродегенеративное заболевание с летальным исходом из группы лизосомных болезней накопления, которое развивается на фоне дефицита фермента лизосом пальмитоил-тио-эстеразы и наследуется по аутосомно-рецессивному типу. Наиболее распространено в Финляндии, где и было описано.
Болезнь Огучи , которая также называется врожденной ночной слепотой , является аутосомно-рецессивной формой врождённой ночной слепоты, связанной с обесцвечиванием глазного дна и аномально медленной адаптацией к темноте.

Бета-маннозидоз — редкое аутосомно-рецессивное наследственное заболевание из группы лизосомных болезней накопления, связанное с нарушением метаболизма олигосахаридов в результате снижения активности фермента лизосом бета-маннозидазы.
Анри́-Жери́ Э́рс — бельгийский физиолог и биохимик, профессор Лувенского католического университета.