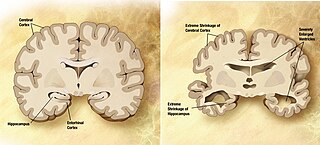
Боле́знь Альцгеймера — наиболее распространённая форма деменции, нейродегенеративное заболевание, впервые описанное в 1907 году немецким психиатром Алоисом Альцгеймером (1864—1915). Как правило, она обнаруживается у людей старше 65 лет, но существует и ранняя болезнь Альцгеймера — редкая форма заболевания. Общемировая заболеваемость на 2006 год оценивалась в 26,6 млн человек, а к 2050 году число больных может вырасти вчетверо.
Боково́й (латера́льный) амиотрофи́ческий склеро́з (БАС; также известен как боле́знь мото́рных нейро́нов, мотонейро́нная боле́знь, боле́знь Шарко́, в англоязычных странах — болезнь Лу Ге́рига — прогрессирующее, неизлечимое дегенеративное заболевание центральной нервной системы, при котором происходит поражение как верхних, так и нижних двигательных нейронов, что приводит к параличам и последующей атрофии мышц.
Болезнь Вильсона — Коновалова — врождённое или приобретенное нарушение метаболизма меди, приводящее к тяжелейшим поражениям центральной нервной системы и внутренних органов. Диагностируется у 5-10 % больных циррозом печени дошкольного и школьного возраста. Заболевание передаётся по аутосомно-рецессивному типу. Ген ATP7B, мутации которого вызывают заболевание, расположен на 13-й хромосоме.

Пресенилины — семейство трансмембранных белков, составляющих часть протеазного комплекса γ-секретазы. В геноме позвоночных содержатся два гена, кодирующих пресенилины: PSEN1 кодирует пресенилин 1, а PSEN2 — пресенилин 2. У человека эти гены расположены, соответственно, на 14-й и 1-й хромосомах. Оба гена эволюционно консервативны — отмечены лишь небольшие отличия между пресенилинами крысы и человека. В организме нематоды Caenorhabditis elegans также существуют два белка, напоминающие пресенилины и, судя по всему, выполняющие сходные функции, — sel-12 и hop-1.
Пресенилин-1 - один из двух известных белков-пресенилинов человека. У некоторых больных семейной болезнью Альцгеймера отмечаются мутации PSEN1 и PSEN2. В исследованиях эти мутации усиливают синтез бета-амилоида, основного составляющего амилоидных бляшек, обнаруживаемых в мозге больных при посмертном анализе. Альтернативный сплайсинг порождает несколько форм пресенилина-1.

Инозитолтрифосфат (IP3) — это водорастворимый вторичный посредник. IP3 образуется в результате распада мембранных фосфолипидов под действием фермента фосфолипазы С. Инозитолтрифосфат вместе с диацилглицерином принимает участие в передаче сигнала в клетке.

Губчатая энцефалопатия крупного рогатого скота, ГЭКРС, коровье бешенство — нейродегенеративная прионная болезнь, приводящая к необратимым, летальным изменениям в головном мозге заражённых животных, относится к группе трансмиссивных губчатых энцефалопатий. Вызывается прионом ГЭКРС. Инкубационный период от 30 месяцев до 8 лет. Передаётся при употреблении в пищу мяса больных животных, вызывает скрейпи у овец и болезнь Крейцфельда-Якоба у людей.

TGFBI — белок человека. Экспрессия TGFBI индуцируется фактором роста TGF-beta. Альтернативное название кератоэпителин свидетельствует об узко выраженной картине экспрессии белка в тканях глаза: он производится клетками эпителия роговицы и стромальными кератоцитами. Ген был впервые описан в 1997 году, изначально получив название BIGH3.

Синдро́м Си́ппла — включает медуллярную карциному щитовидной железы, двустороннюю (билатеральную) феохромоцитому и первичный гиперпаратиреоз. Возможно сочетание с амилоидозом кожи или болезнью Гиршпрунга.

Точечная мутация — тип мутации в ДНК или РНК, при котором одно азотистое основание заменяется другим. Термин также применяется и в отношении парных замен, инсерции или делеции одного или нескольких нуклеотидов. Точечные мутации, возникающие в некодирующей ДНК, обычно никак себя не проявляют. Точечный мутант — организм, в генотипе которого произошла точечная мутация.
Мутация сдвига рамки считывания — тип мутации в последовательности ДНК, для которого характерна вставка или делеция нуклеотидов, в количестве не кратном трём. В результате происходит сдвиг рамки считывания при транскрипции мРНК. Мутации сдвига рамки считывания делятся на мишенные, немишенные, мишенные задерживающиеся и немишенные задерживающиеся мутации сдвига рамки считывания.

Нейрофиброматоз I (первого) типа — самое распространённое наследственное заболевание, предрасполагающее к возникновению опухолей у человека. Описан во второй половине XIX века рядом исследователей, в том числе в 1882 году учеником Рудольфа Вирхова Фридрихом фон Реклингхаузеном. Устаревшие названия — болезнь Реклингхаузена, периферический нейрофиброматоз и др. Является аутосомно-доминантным, встречается с одинаковой частотой у мужчин и у женщин, у 1 из 3500 новорождённых. Другие типы нейрофиброматозов характеризуются наличием как сходных проявлений с I типом, так и отличий.

Острый мегакариобластный лейкоз (ОМКЛ) — это такая форма острого миелоидного лейкоза, при которой большинство лейкозных бластных клеток представляют собой мегакариобласты.

Боле́знь Фабри́ — редкое генетически детерминированное заболевание с Х-сцепленным типом наследования, из группы лизосомных болезней накопления. Ранее считалось, что тип наследования болезни Фабри — X-сцепленный рецессивный, однако на современном этапе накоплено достаточно данных, чтобы считать тип наследования болезни Фабри X-сцепленным доминантным с неполной пенетрантностью у женщин. Данное заболевание вызвано нарушением метаболизма сфинголипидов и обладает широким спектром клинических симптомов.

GM2-ганглиозидо́з — тяжёлое наследственное заболевание, развивающееся в результате дефицита или недостаточной активности фермента гексозаминидазы и накопления в клетках ганглиозидов. Относится к лизосомным болезням накопления и имеет три варианта, связанные с мутациями в разных генах, которые оказывают вляние на активность общей гексозаминидазы. Два варианта заболевания больше известны под индивидуальными именами, полученными в честь авторов, впервые описавших их клиническую картину: болезнь Тея — Сакса, болезнь Сандхоффа. Третий вариант этой болезни носит название GM2 ганглиозидоз, вариант АБ. Болезнь Тея — Сакса вызвана мутацией в гене HEXA, кодирующий альфа-субъединицу гексоаминидазы А. Болезнь Сандхоффа вызвана мутацией в гене HEXB, кодирующий бета-субъединицу гексоаминидаз А и Б. GM2 ганглиозидоз, АБ вариант связан с нарушением в гене GM2, кодирующий белок-активатор GM2A.
GM2 ганглиозидо́з, вариа́нт АБ (англ. GM2-gangliosidosis, AB variant) — редкое аутосомно-рецессивное нарушение обмена веществ, связанное с мутацией гена GM2A. Характеризуется нормальной активностью β-гексозаминидаз А и Б и обусловлено недостаточностью активатора (белкового кофактора), необходимого для реализации ферментной активности в отношении субстрата. Заболевание клинически проявляется прогрессирующим разрушением нервных клеток головного и спинного мозга.
Сиалолипидо́з — аутосомно-рецессивное наследственное заболевание из группы муколипидозов, относящееся к лизосомным болезням накопления. Клиническая картина отличается разнообразной симптоматикой, включающей задержку психомоторного развития и различные глазные аберрации. Заболевание обусловлено различными мутациями гена MCOLN1, расположенного на длинном плече 19-й хромосомы (19q13.3-p13.2), который кодирует неселективный катионный канал муколипина-1. Результатом данной генной мутации является нарушение клеточных функций, которое ведёт к патологии нервной системы посредством неизвестного механизма.

Холин-О-ацетилтрансфераза, также холин-ацетилтрансфераза, холинацетил-СоА-трансфераза (англ. Choline acetyltransferase, сокр. СhAT, ХАТ, но иногда и CAT) — фермент (КФ 2.3.1.6), из семейства ацилтрансфераз (класс трансферазы), катализирующий реакцию переноса ацетильной группы (CH3-CO) от молекулы ацетил-CoA на молекулу субстрата — холина, с образованием ацетилхолина (АЦХ) и кофермента А, по уравнению:
- ацетил-СоА + холин
 ацетилхолин + CoA-SH.
ацетилхолин + CoA-SH.

Болезнь Шарко — Мари — Тута (ШМТ), или наследственная моторно-сенсорная нейропатия (НМСН) — наследственная периферическая нейропатия с хроническим прогрессирующим течением. При этой болезни больные страдают от слабости и атрофии мышц дистальных отделов конечностей, деформации стоп и кистей, у них наблюдается снижение сухожильных рефлексов, изменение походки, потеря чувствительности в конечностях. В основе клинических проявлений болезни лежит поражение двигательных и чувствительных периферических нервных волокон. Болезнь Шарко — Мари — Тута диагностируется с приблизительной частотой 1 на 2500 человек. Первое проявление болезни чаще всего происходит в подростковом возрасте или в раннем взрослом возрасте. Тяжесть симптомов сильно различается даже среди членов одной семьи с этим заболеванием. Болезнь Шарко — Мари — Тута нередко ведёт к ограничению трудоспособности и к инвалидизации, при этом большинство пациентов имеют нормальную продолжительность жизни. Болезнь Шарко — Мари — Тута является генетически крайне неоднородным заболеванием, симптомы этой болезни могут быть вызваны мутациями в более чем двух десятках генов, хотя большая часть заболеваний вызвана мутациями в генах PMP22, MPZ, GJB1 и MFN2. Наследование болезни чаще всего аутосомно-доминантное, однако может быть аутосомно-рецессивным и Х-сцепленным.
Ранняя болезнь Альцгеймера, также болезнь Альцгеймера с ранним началом — болезнь Альцгеймера, диагностируемая в возрасте до 65 лет. Это необычная форма болезни, на которую приходится всего 5—10 % всех случаев заболеваний. Около 60 % имеют положительный семейный анамнез болезни Альцгеймера, и 13 % из них наследуются по аутосомно-доминантному типу. Большинство случаев болезни Альцгеймера с ранним началом имеют те же черты, что и обычная форма, и не вызваны известными генетическими мутациями.