
Синдром Ретта — психоневрологическое наследственное заболевание, встречается почти исключительно у девочек с частотой 1:10000 — 1:15000, является причиной тяжёлой умственной отсталости у девочек.

Синдром Ангельмана — обусловленная генетической аномалией патология, характеризующаяся такими признаками, как задержка психического развития, нарушения сна, припадки, хаотические движения, частый смех или улыбки. Также эту болезнь называют «синдром Петрушки» или «синдром счастливой куклы».

Синдро́м Ма́ртина — Белл — наследственное заболевание.

Синдро́м Альстрёма — генетическая патология человека, относящаяся к группе цилиопатий. Характеризуется пигментной дегенерацией сетчатки, ожирением, прогрессирующей нейросенсорной глухотой, дилятационной кардиомиопатией, сахарным диабетом и нефропатией. Впервые описан в 1959 году шведским психиатром Карлом-Генри Альстрёмом.
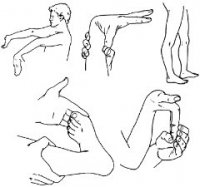
Синдром Э́лерса — Данло́са — это группа наследственных системных дисфункций соединительной ткани, вызванных дефектом в синтезе коллагена. В зависимости от отдельной мутации, серьёзность синдрома может измениться от умеренного до опасного для жизни. Лечения нет, но существует терапия (уход), смягчающая последствия.

Дисплазия соединительной ткани — системное заболевание соединительной ткани, генетически гетерогенное и клинически полиморфное патологическое состояние, обусловленное нарушением развития соединительной ткани в эмбриональном и постнатальном периодах. Характеризуется дефектами волокнистых структур и основного вещества соединительной ткани, приводящее к расстройству гомеостаза на тканевом, органном и организменном уровнях в виде различных морфофункциональных нарушений висцеральных и локомоторных органов с прогредиентным течением. Синонимы: соединительнотканная дисплазия, наследственное нарушение соединительной ткани, врожденная соединительнотканная недостаточность, системное невоспалительное заболевание соединительной ткани, гипермобильный синдром, наследственная коллагенопатия.
Цилиопатии — генетически обусловленные заболевания, возникающие при нарушении структуры или функции цилий.

Синдром MERRF — редкое митохондриальное заболевание, вызываемое мутациями в следующих генах: MTTK, MTTL1, MTTH, MTTS1, MTTS2, MTTF. Симптомы MERRF также проявляются при мутациях гена MTND5.
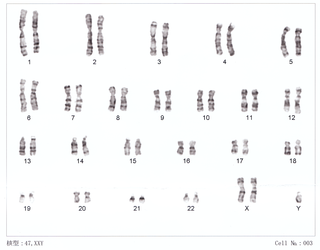
Синдром Клайнфельтера — генетическое отклонение. Клиническая картина синдрома описана в 1942 году в работах Гарри Клайнфельтера и Фуллера Олбрайта. Генетической особенностью этого синдрома является разнообразие цитогенетических вариантов и их сочетаний (мозаицизм). Обнаружено несколько типов полисомии по хромосомам X и Y у лиц мужского пола: 47, XXY; 47, XYY; 48, XXXY; 48, XYYY; 48 XXYY; 49 XXXXY; 49 XXXYY. Наиболее распространён синдром Клайнфельтера. Общая частота его колеблется в пределах 1 на 500—700 новорождённых мальчиков, что делает данный синдром первым по частоте встречаемости среди хромосомных болезней.

Синдро́м Нуна́н — редкая врождённая патология, как правило, наследуется по аутосомно-доминантному типу, носит семейный характер, однако встречаются и спорадически. Синдром предполагает наличие фенотипа, характерного для синдрома Шерешевского-Тернера у особей женского и мужского пола с нормальным генотипом.
Синдро́м Шми́дта — включает недостаточность надпочечников, лимфоцитарный тиреоидит, гипопаратиреоз и недостаточность половых желез в любом сочетании этих симптомов друг с другом; также возможен сахарный диабет первого типа. Синдром назван в честь Schmidt A., впервые описавшего данный симптомокомплекс в 1926 году. Является наиболее распространённым типом синдрома полигландулярной недостаточности. Чаще болеют женщины.
Аутоимму́нный полиэндокри́нный синдро́м — аутоиммунное заболевание, объединяющее поражение нескольких эндокринных органов.
Синдро́м де ля Шапе́ля — назван в честь исследователя, впервые охарактеризовавшего его в 1972 году. Синдром относится к редкой хромосомной патологии, возникающей в результате кроссинговера между X- и Y-хромосомами в процессе мейоза, в результате чего одна или обе X-хромосомы содержат нормальный мужской ген SRY. Распространённость данного синдрома составляет 4—5 на 100 000, что меньше встречаемости синдрома Клайнфельтера.
Синдро́м Ме́ккеля — Гру́бера — генетическая патология человека, относящаяся к группе цилиопатий. Впервые описан немецким врачом Иоганном Меккелем в 1822 году.
Втори́чные фо́рмы са́харного диабе́та — разнородная группа заболеваний, к которой относится сахарный диабет, встречающийся на фоне другой клинической патологии, которая может и не сочетаться с сахарным диабетом. Для большинства заболеваний из этой группы этиологические факторы раскрыты. Кроме того, к этой группе заболеваний также относят и некоторые генетические (наследственные) синдромы, в том числе аномалии инсулиновых рецепторов. При вторичных формах сахарного диабета отсутствуют ассоциации с HLA-антигенами, данные за аутоиммунное поражение и антитела к островковой ткани поджелудочной железы.
Синдром Неймегена является редким аутосомно-рецессивным наследственным синдромом хромосомной нестабильности, при котором наблюдается микроцефалия, иммунодефицит и склонность к злокачественным новообразованиям. Синдром Неймегена вызван мутациями в гене NBN, который участвует в клеточном ответе на повреждения ДНК.
Синдром Коккейна , также называемый синдром Нил-Дингуолл — редкое аутосомно-рецессивное, нейродегенеративное расстройство, характеризующееся недостатком роста, нарушением развития нервной системы, аномальной чувствительностью к солнечному свету (фотосенсибилизация), заболеваниями глаз и преждевременным старением. Нездоровый вид и неврологические расстройства являются критериями для диагностики, а светочувствительность, нарушения слуха и ненормальные глаза — другие весьма общие черты. Возможны проблемы любого или всех внутренних органов. Это связано с группой расстройств, называемых лейкодистрофия. В основе расстройства лежит дефект механизма репарации ДНК. Интересно, в отличие от других дефектов репарации ДНК, пациенты с CS не предрасположены к раку или инфекции. Синдром Коккейна редок, но это разрушительная болезнь, которая, как правило, приводит к смерти в первом или втором десятилетии жизни. Мутация специфических генов в синдроме Коккейна известна, но широко распространенные эффекты и его отношения с репарацией ДНК еще не очень хорошо исследованы.
Синдром Пирсона — тяжелое врожденное аутосомно-рецессивное заболевание, вызванное мутацией гена, ответственного за кодирование β2-цепи ламинина. На долю которого приходится около 2,5% нефротического синдрома в течение первого года жизни, обычно клинически проявляющегося в течение первых 3 месяцев. В России зафиксирован единичный случай этого заболевания.
Синдром Смит — Магенис (СМС) — наследственное заболевание, имеет такие проявления, как умственная отсталость, аномалии лица, проблемы со сном и многочисленные поведенческие проблемы, в том числе и такие, как самоповреждение. Частота синдрома Смит — Магенис составляет 1 случай на 15 000-25 000 человек. Синдром Смит — Магенис, как правило, обусловлен микроделецией в коротком плече 17-й хромосомы (17p), и иногда его называют 17p-синдром.
Мутация CDKL5 — редкое генетическое заболевание, обнаруженное на X-хромосоме. Впервые было выявлено в 2004 году..

