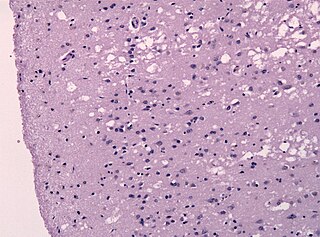
Прио́ны — особый класс инфекционных патогенов, не содержащих нуклеиновых кислот. Прионы представляют собой белки с аномальной третичной структурой. Это положение лежит в основе прионной гипотезы, однако насчёт состава прионов существуют и другие точки зрения.

Ку́ру — редкое неизлечимое смертельное нейродегенеративное прионное заболевание, встречающееся в высокогорных районах Новой Гвинеи у аборигенов племени форе. Представляет собой форму трансмиссивной губчатой энцефалопатии (TSE), вызванную передачей аномально свернутых белков (прионов), что приводит к таким симптомам, как тремор и потеря координации из-за нейродегенерации. Впервые обнаружено в начале XX века.

Деме́нция — приобретённое слабоумие, стойкое снижение познавательной деятельности с утратой в той или иной степени ранее усвоенных знаний и практических навыков и затруднением или невозможностью приобретения новых. В отличие от умственной отсталости, представляющей собой недоразвитие психики, деменция — это распад психических функций, происходящий в результате заболевания или повреждения головного мозга после завершения его созревания. Наиболее часто деменция наблюдается в старости. В народе сенильная деменция носит название ста́рческий мара́зм. По данным ВОЗ, в 2015 году во всём мире насчитывалось более 46 миллионов людей с деменцией. В 2017 году это число увеличилось до 50 миллионов. Ежегодно регистрируются 7,7 миллиона новых случаев деменции, каждый из которых становится тяжким бременем для семей и систем здравоохранения. Ожидается, что это число увеличится до 131,5 млн к 2050 году.

Кардиомиопатии — гетерогенная группа заболеваний миокарда, связанных с механической или электрической дисфункцией, которая обычно проявляется неадекватной гипертрофией или дилатацией. Кардиомиопатии могут как изолированно поражать только сердце, так и быть частью генерализованного системного заболевания, часто приводят к сердечно-сосудистой смерти или к инвалидизации, обусловленной прогрессирующей сердечной недостаточностью[].

Фатальная семейная бессонница — редкое неизлечимое наследственное, нейродегенеративное (доминантно-наследуемое) прионное заболевание, при котором больной неизбежно умирает от бессонницы. Известно всего 42 семьи, поражённых этой болезнью.

Болезнь Кре́йтцфельдта — Я́коба, — прогрессирующее дистрофическое заболевание коры большого мозга, базальных ганглиев и спинного мозга. Считается основным проявлением губчатой энцефалопатии. Смерть наступает в 100% случаев.
Энцефалопа́тия и энце́фалопати́я, также органи́ческое пораже́ние головно́го мо́зга — общее название для невоспалительных заболеваний головного мозга, при которых дистрофически изменяется ткань мозга, что приводит к нарушению его функции. Энцефалопатия бывает врождённая и приобретённая.
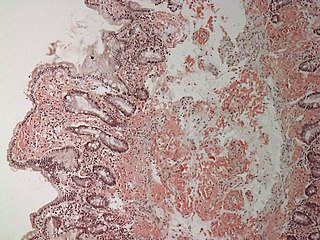
Амилоидóз — нарушение белкового обмена, сопровождающееся образованием и отложением в тканях специфического белково-полисахаридного комплекса — амилоида.

Синдро́м Ре́я, синдром Ре́йе — редкое, но очень опасное, часто угрожающее жизни острое состояние, возникающее у детей и подростков на фоне лечения лихорадки вирусного происхождения препаратами, содержащими ацетилсалициловую кислоту, и характеризующееся быстро прогрессирующей энцефалопатией и развитием жировой инфильтрации печени. Синдром Рея сопровождается гипераммониемией, повышением уровня АСТ, АЛТ в сыворотке крови при нормальном уровне билирубина.
Болезнь Вильсона — Коновалова — врождённое или приобретенное нарушение метаболизма меди, приводящее к тяжелейшим поражениям центральной нервной системы и внутренних органов. Диагностируется у 5-10 % больных циррозом печени дошкольного и школьного возраста. Заболевание передаётся по аутосомно-рецессивному типу. Ген ATP7B, мутации которого вызывают заболевание, расположен на 13-й хромосоме.
Атаксия Фридрейха — аутосомно-рецессивное заболевание, характеризующееся дегенеративным повреждением нервной системы вследствие наследуемой мутации в гене FXN, кодирующем белок фратаксин. Заболевание наследуется по аутосомно-рецессивному типу. Заболевание названо в честь немецкого врача Николауса Фридрейха, который первым описал её в 1860 году.

Губчатая энцефалопатия крупного рогатого скота, ГЭКРС, коровье бешенство — нейродегенеративная прионная болезнь, приводящая к необратимым, летальным изменениям в головном мозге заражённых животных, относится к группе трансмиссивных губчатых энцефалопатий. Вызывается прионом ГЭКРС. Инкубационный период от 30 месяцев до 8 лет. Передаётся при употреблении в пищу мяса больных животных, вызывает скрейпи у овец и болезнь Крейцфельда-Якоба у людей.

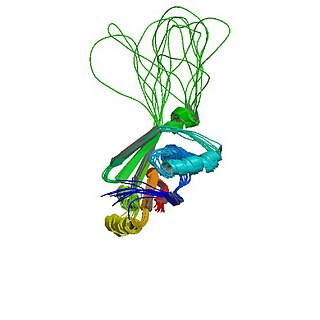
PRNP — ген, расположенный на коротком плече 20-й хромосомы. Кодирует нормальный прионный белок (PrPC) и изоформу этого белка — прионный белок PrPSc, связанный с заболеваниями. Пока не ясно, как именно PrPC превращается в PrPSc, как реплицируется PrPSc и почему его накопление вызывает дегенерацию нейронов.
Синдром Альперса — наследственная болезнь, характеризующаяся развитием у детей слепоты, локальных или генерализованных судорог, миоклонических проявлений, слабоумия. Течение очень быстрое, с образованием слабоумия. На сегодняшний день заболевание неизлечимое, и чем младше больной, тем быстрее наступает смерть.
Периодическая болезнь — сравнительно редкое, генетически обусловленное наследственное заболевание, проявляющееся периодически рецидивирующим серозитом и относительно частым развитием амилоидоза. Встречается преимущественно у представителей народностей, предки которых жили в странах на территории Средиземноморского бассейна, особенно у армян, евреев, арабов и лишь в 6% случаев у лиц прочих национальностей.

Синдро́м Си́ппла — включает медуллярную карциному щитовидной железы, двустороннюю (билатеральную) феохромоцитому и первичный гиперпаратиреоз. Возможно сочетание с амилоидозом кожи или болезнью Гиршпрунга.

Деменция с тельцами Леви : предположительно является второй после болезни Альцгеймера по частоте среди нейродегенеративных деменций. Согласно нейропатологическим исследованиям, деменция с тельцами Леви (ДТЛ) определялась в приблизительно 30 % случаев всех деменций.
Нейродегенеративные заболевания — группа в основном медленно прогрессирующих, наследственных или приобретённых заболеваний нервной системы. Общим для этих заболеваний является прогрессирующая гибель нервных клеток (нейродегенерация), ведущая к различным неврологическим симптомам — прежде всего, к деменции и нарушению движений. Заболевания могут наступить в различном возрасте, протекают диффузно или генерализированно, гистологически определяется специфический тип изменений.
Трансмиссивные губчатые энцефалопатии (ТГЭ), или трансмиссивные спонгиоформные энцефалопатии (ТСЭ), также известные как прионные болезни — группа нейродегенеративных заболеваний людей и животных c образованием губчатой энцефалопатии, которая принадлежит к медленным инфекциям и характеризуется поражением центральной нервной системы, мышечной, лимфатической и других систем, с летальным исходом.
Ранняя болезнь Альцгеймера, также болезнь Альцгеймера с ранним началом — болезнь Альцгеймера, диагностируемая в возрасте до 65 лет. Это необычная форма болезни, на которую приходится всего 5—10 % всех случаев заболеваний. Около 60 % имеют положительный семейный анамнез болезни Альцгеймера, и 13 % из них наследуются по аутосомно-доминантному типу. Большинство случаев болезни Альцгеймера с ранним началом имеют те же черты, что и обычная форма, и не вызваны известными генетическими мутациями.
