
Насле́дственные заболева́ния — заболевания, возникновение и развитие которых связано с различными дефектами и нарушениями в наследственном аппарате клеток. В основе наследственных заболеваний лежат мутации: хромосомные, генные и митохондриальные. Наследственные заболевания могут быть обусловлены мутациями, передаваемыми в семьях по наследству, или мутациями, вновь возникшими в клетках зародышевой линии, в зиготе или на очень ранних этапах развития. Наследственные болезни многочисленны и разнообразны по проявлениям.

Синдром (болезнь) Марфана аутосомно-доминантное заболевание из группы наследственных патологий соединительной ткани. Синдром вызван мутациями генов, кодирующих синтез гликопротеина фибриллина-1, и является плейотропным. Заболевание характеризуется различной пенетрантностью и экспрессивностью. В классических случаях лица с синдромом Марфана высоки (долихостеномелия), имеют удлинённые конечности, вытянутые пальцы (арахнодактилия) и недоразвитие жировой клетчатки. Помимо характерных изменений в органах опорно-двигательного аппарата, наблюдается патология в органах зрения и сердечно-сосудистой системы, что в классических вариантах составляет триаду Марфана.

Синдро́м Альстрёма — генетическая патология человека, относящаяся к группе цилиопатий. Характеризуется пигментной дегенерацией сетчатки, ожирением, прогрессирующей нейросенсорной глухотой, дилятационной кардиомиопатией, сахарным диабетом и нефропатией. Впервые описан в 1959 году шведским психиатром Карлом-Генри Альстрёмом.
Синдро́м (болезнь) де То́ни — Дебре́ — Фанко́ни — врождённое заболевание, наследуется по аутосомно-рецессивному типу. Комплекс биохимических и клинических проявлений поражения проксимальных почечных канальцев с нарушением канальцевой реабсорбции фосфата, глюкозы, аминокислот и бикарбоната. Одно из рахитоподобных заболеваний.

Синдром Ваарденбурга — наследственное заболевание. Имеет следующие клинические признаки: телекант, гетерохромия радужки, седая прядь надо лбом и врождённая тугоухость различной степени.
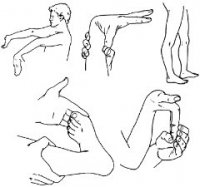
Синдром Э́лерса — Данло́са — это группа наследственных системных дисфункций соединительной ткани, вызванных дефектом в синтезе коллагена. В зависимости от отдельной мутации, серьёзность синдрома может измениться от умеренного до опасного для жизни. Лечения нет, но существует терапия (уход), смягчающая последствия.

Дисплазия соединительной ткани — системное заболевание соединительной ткани, генетически гетерогенное и клинически полиморфное патологическое состояние, обусловленное нарушением развития соединительной ткани в эмбриональном и постнатальном периодах. Характеризуется дефектами волокнистых структур и основного вещества соединительной ткани, приводящее к расстройству гомеостаза на тканевом, органном и организменном уровнях в виде различных морфофункциональных нарушений висцеральных и локомоторных органов с прогредиентным течением. Синонимы: соединительнотканная дисплазия, наследственное нарушение соединительной ткани, врожденная соединительнотканная недостаточность, системное невоспалительное заболевание соединительной ткани, гипермобильный синдром, наследственная коллагенопатия.
Синдром Лея или подострая некротизирующая энцефаломиопатия — редкий наследственный нейрометаболический синдром, поражающий ЦНС. На сегодняшний день лечения данного заболевания не известно. У синдрома множество причин, значительную роль играют мутации генов, связанных с работой митохондрий. В сентябре 2016 года появилось сообщение о первой профилактике заболевания путём рождения ребёнка «от трёх родителей» — с использованием митохондрий донорской яйцеклетки.

Синдром MERRF — редкое митохондриальное заболевание, вызываемое мутациями в следующих генах: MTTK, MTTL1, MTTH, MTTS1, MTTS2, MTTF. Симптомы MERRF также проявляются при мутациях гена MTND5.

Синдром Робинова. Редкое генетическое заболевание, для которого на общем фоне отставания в развитии характерны — увеличенная голова, выпуклый лоб, низкий рост, полнота, брахидактилия конечностей, неразвитость внешних половых органов и др. Впервые синдром был описан в 1969 году в американском журнале заболеваний у детей педиатром и генетиком Мейнхардом Робиновым вместе с коллегами Фредериком Сильверманом и Хьюго Смитом. К 2002 году изучено и опубликовано в специализированной медицинской литературе более 100 клинических случаев заболевания. Синдром также известен как Робинов-Сильверман-Смит синдром. Рецессивная форма заболевания была ранее известна как синдром Covesdem.

DIDMOAD-синдро́м — аутосомно-рецессивно наследуемый синдром, ассоциированный с инсулинозависимым сахарным диабетом и прогрессирующей атрофией диска зрительного нерва, которые выявляют до 16-летнего возраста. Сочетается с двусторонней прогрессирующей нейросенсорной тугоухостью, несахарным диабетом центрального генеза, дисфункцией автономной нервной системы, приводящей к развитию нейропатического мочевого пузыря и другим проявлениям нейродегенерации, включающими мозжечковую атаксию, миоклональную эпилепсию и атрофию ствола головного мозга. Развёрнутая клиническая картина (фенотипически) встречается приблизительно у 75 % пациентов. Сахарный диабет неаутоиммунного генеза, клинические проявления недостаточности инсулина проявляются приблизительно в 6-летнем возрасте. Средняя продолжительность жизни достигает 30 лет, в течение этого срока происходит развитие полного фенотипа данного синдрома.

Синдро́м Нуна́н — редкая врождённая патология, как правило, наследуется по аутосомно-доминантному типу, носит семейный характер, однако встречаются и спорадически. Синдром предполагает наличие фенотипа, характерного для синдрома Шерешевского-Тернера у особей женского и мужского пола с нормальным генотипом.
Синдро́м Ме́ккеля — Гру́бера — генетическая патология человека, относящаяся к группе цилиопатий. Впервые описан немецким врачом Иоганном Меккелем в 1822 году.

Синдро́м тетраамели́и — очень редкое врождённое наследственное заболевание, характеризующееся отсутствием четырёх конечностей. Другие части тела, такие как лицо, череп, репродуктивные органы, анус и таз, также подвержены порокам развития. Для синдрома тетраамелии характерно аутосомно-рецессивное наследование. Заболевание связано с мутацией в гене WNT3.

Нейродегенерация с отложением железа в мозге или Болезнь Галлервордена — Шпатца — очень редкое нейродегенеративное заболевание, сопровождающееся отложением железа в базальных ганглиях. Это аутосомно-рецессивно наследуемое заболевание было впервые описано в 1922 году Юлиусом Галлеворденом и Гуго Шпатцем. Частота заболевания в среднем 1-3 человека на 1 миллион.

X-сцепленное рецессивное наследование — один из видов сцепленного с полом наследования. Такое наследование характерно для признаков, гены которых расположены на Х-хромосоме и которые проявляются только в гомозиготном или гемизиготном состоянии. Такой тип наследования имеет ряд врождённых наследственных заболеваний у человека, эти заболевания связаны с дефектом какого-либо из генов, расположенных на половой Х-хромосоме, и проявляются в случае, если нет другой Х-хромосомы с нормальной копией того же гена. В литературе встречается сокращение XR для обозначения X-сцепленного рецессивного наследования.
Синдро́м Ке́рнса — Се́йра (англ. Kearns–Sayre syndrome, сокращённо KSS) — митохондриальная миопатия с типичным началом до 20-летнего возраста. KSS является более серьёзным синдромным вариантом хронической прогрессирующей внешней офтальмоплегии, синдром, который характеризуется изолированным поражением мышц, контролирующих движения век и контролирующих движения глаз. Это приводит к птозу и офтальмоплегии соответственно. KSS включает в себя триаду уже описанных CPEO, двустороннюю пигментную ретинопатию и блокаду сердца. Другие области участия могут включать в себя мозжечковую атаксию, проксимальную мышечную слабость, глухоту, сахарный диабет, дефицит гормона роста, гипопаратиреоз или другие эндокринные нарушения. В обоих этих заболеваниях вовлечение мышц может начаться односторонним, но всегда развивается в двусторонний дефицит, который является прогрессирующим.
Синдром Неймегена является редким аутосомно-рецессивным наследственным синдромом хромосомной нестабильности, при котором наблюдается микроцефалия, иммунодефицит и склонность к злокачественным новообразованиям. Синдром Неймегена вызван мутациями в гене NBN, который участвует в клеточном ответе на повреждения ДНК.
Синдром Коккейна , также называемый синдром Нил-Дингуолл — редкое аутосомно-рецессивное, нейродегенеративное расстройство, характеризующееся недостатком роста, нарушением развития нервной системы, аномальной чувствительностью к солнечному свету (фотосенсибилизация), заболеваниями глаз и преждевременным старением. Нездоровый вид и неврологические расстройства являются критериями для диагностики, а светочувствительность, нарушения слуха и ненормальные глаза — другие весьма общие черты. Возможны проблемы любого или всех внутренних органов. Это связано с группой расстройств, называемых лейкодистрофия. В основе расстройства лежит дефект механизма репарации ДНК. Интересно, в отличие от других дефектов репарации ДНК, пациенты с CS не предрасположены к раку или инфекции. Синдром Коккейна редок, но это разрушительная болезнь, которая, как правило, приводит к смерти в первом или втором десятилетии жизни. Мутация специфических генов в синдроме Коккейна известна, но широко распространенные эффекты и его отношения с репарацией ДНК еще не очень хорошо исследованы.
Синдро́м Ло́уренса-Му́на. Редкое аутосомно-рецессивное генетическое заболевание, характеризующиеся ожирением, умственной отсталостью, атаксией, дистрофией сетчатки, гипопитуитаризмом, большим количеством пальцев рук или ног. Синдром связан с нарушением функции гипоталамических центров.