
Синдро́м Туре́тта — генетически обусловленное расстройство центральной нервной системы, которое проявляется в любом возрасте и характеризуется множественными двигательными («моторными») тиками и как минимум одним голосовым, появляющимися много раз в течение дня. В американском Диагностическом и статистическом руководстве по психическим расстройствам пятого издания (DSM-5), наряду с остальными тикозными расстройствами, относится к нейроонтогенетическим моторным расстройствам. Довольно часто встречается коморбидное состояние: синдром Туретта с синдромом дефицита внимания и гиперактивности.

Синдро́м Аспе́ргера — общее нарушение психического развития, характеризующееся серьёзными трудностями в социальном взаимодействии, а также ограниченным, стереотипным, повторяющимся репертуаром интересов и занятий. От детского аутизма он отличается прежде всего тем, что речевые и когнитивные способности в целом сохраняются. Синдром часто характеризуется также выраженной неуклюжестью.
Синдром поликистозных яичников — полиэндокринный синдром, сопровождающийся нарушениями функции яичников, поджелудочной железы, коры надпочечников, гипоталамуса и гипофиза.

Мыс вдовы — линия роста волос на лбу в форме треугольника вершиной вниз. Признак наследуется генетически и является доминантным сцепленным с X-хромосомой.

Синдро́м Праде́ра — Ви́лли — редкое наследственное заболевание, причиной которого является отсутствие отцовской копии участка хромосомы 15q11-13. В этом участке хромосомы 15 находятся гены, в регуляции которых задействован геномный импринтинг. Большинство случаев являются спорадическими, для редких описанных семейных случаев характерно неменделевское наследование.
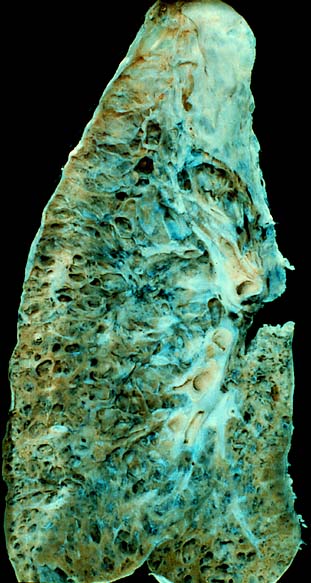
Интерстициальные заболевания легких (ИЗЛ) — преимущественно хронические заболевания лёгочной ткани, проявляющиеся воспалением и нарушением структуры альвеолярных стенок, эндотелия лёгочных капилляров, перивазальных и перилимфатических тканей. Характерным симптомом интерстициальных заболеваний лёгких является одышка, являющаяся отражением лёгочной недостаточности.
Синдром Фрейзера — аутосомно-рецессивное врождённое заболевание. Синдром назван по имени медицинского генетика Джорджа Фрейзера.

Синдро́м Шереше́вского — Тёрнера (СШТ) — хромосомный симптомокомплекс, обусловленный частичной или полной моносомией по X-хромосоме, в то время как здоровые женщины имеют и унаследованную от матери хромосому, и хромосому от отца. Характеризуется аномалиями физического развития, низкорослостью, половым инфантилизмом и другими разнящимися признаками. Традиционным методом верифицирования является кариотипирование. Терапия основана на приёме гормонов роста и эстрогенов.

Множественная эндокринная неоплазия типа IIb — в данный синдром вошли те же опухоли, что и в МЭН IIa, однако первичный гиперпаратиреоз проявляется реже, а медуллярная карцинома щитовидной железы протекает крайне злокачественно. Выявляется в возрасте 4—37 лет. Отличается от МЭН IIa наличием нейрином (невром) слизистых оболочек и расстройств опорно-двигательного аппарата. Нейриномы чаще локализуются на губах, щеках, языке, других отделах желудочно-кишечного тракта и представляют собой бело-розовые безболезненные узелки диаметром от 1—3 мм до 1 см. Множественная эндокринная неоплазия типа IIb является самым серьёзным вариантом синдрома МЭН.

Синдро́м Ларо́на — наследственное заболевание с аутосомно-рецессивным типом наследования — своеобразная разновидность карликовости (низкорослости), обусловленная врождённым дефектом гена рецептора соматотропного гормона (СТГ), приводящим к нечувствительности периферических тканей к действию гормона роста. Резистентность к ИФР-1 выявлена у африканских пигмеев.

Синдро́м Ба́рттера — форма гиперальдостеронизма с гиперплазией юкстагломерулярного аппарата почек и резистентностью к сосудосуживающему действию ангиотензина II, обусловленной внешними (вторичными) нарушениями передачи сигнала ангиотензина II.

Синдро́м Ка́рнея — редкое наследственное заболевание с аутосомно-доминантным типом наследования. Характеризуется образованием у детей множественных опухолей. Следует отличать от триады Карни.
Синдро́м де ля Шапе́ля — назван в честь исследователя, впервые охарактеризовавшего его в 1972 году. Синдром относится к редкой хромосомной патологии, возникающей в результате кроссинговера между X- и Y-хромосомами в процессе мейоза, в результате чего одна или обе X-хромосомы содержат нормальный мужской ген SRY. Распространённость данного синдрома составляет 4—5 на 100 000, что меньше встречаемости синдрома Клайнфельтера.

Синдром Блума или Синдром Блум-Торре-Мачэйкик — редкое аутосомно-рецессивное заболевание, для которого характерны низкий рост больных и предрасположенность к онкологическим заболеваниям. Клетки больных синдромом Блума демонстрируют сильную геномную нестабильность, в частности, частую гомологичную рекомбинацию. Заболевание было обнаружено и впервые описано американским дерматологом доктором Дэвидом Блумом в 1954 году.
Окулярный ишемический синдром — созвездие глазных признаков и симптомов вторичных по отношению к тяжёлой, хронической артериальной гипоперфузии глаза. Амавроз слепоты является формой острой потери зрения, вызванной снижением кровотока в глазе, может стать предупреждающим признаком предстоящего инсульта. Следовательно, страдающим временами нечеткостью зрения, следует срочно обратиться к врачу для тщательной оценки сонной артерии. Передний сегмент ишемического синдрома — ишемическая состояние переднего сегмента обычно наблюдался в пост-хирургических случаях. Артериальная окклюзия сетчатки глаза приводит к быстрой гибели клеток сетчатки, в результате чего — серьезной потере зрения.
Синдром Фостер Кеннеди относится к плеяде находок, связанных с опухолями лобных долей.
Синдро́м Ке́рнса — Се́йра (англ. Kearns–Sayre syndrome, сокращённо KSS) — митохондриальная миопатия с типичным началом до 20-летнего возраста. KSS является более серьёзным синдромным вариантом хронической прогрессирующей внешней офтальмоплегии, синдром, который характеризуется изолированным поражением мышц, контролирующих движения век и контролирующих движения глаз. Это приводит к птозу и офтальмоплегии соответственно. KSS включает в себя триаду уже описанных CPEO, двустороннюю пигментную ретинопатию и блокаду сердца. Другие области участия могут включать в себя мозжечковую атаксию, проксимальную мышечную слабость, глухоту, сахарный диабет, дефицит гормона роста, гипопаратиреоз или другие эндокринные нарушения. В обоих этих заболеваниях вовлечение мышц может начаться односторонним, но всегда развивается в двусторонний дефицит, который является прогрессирующим.
Синдро́м ли́зиса о́пухоли — группа метаболических нарушений, возникающих как осложнение при лечении онкологических заболеваний. Вызывается гибелью большого количества клеток опухоли за короткий период времени, при которой их содержимое попадает в кровь. Чаще всего возникает при лечении лимфом и лейкозов. В онкологии и гематологии это потенциально смертельное осложнение, поэтому пациенты с риском его возникновения должны находиться под наблюдением во время всего периода лечения химиотерапией.
Синдром Смит — Магенис (СМС) — наследственное заболевание, имеет такие проявления, как умственная отсталость, аномалии лица, проблемы со сном и многочисленные поведенческие проблемы, в том числе и такие, как самоповреждение. Частота синдрома Смит — Магенис составляет 1 случай на 15 000-25 000 человек. Синдром Смит — Магенис, как правило, обусловлен микроделецией в коротком плече 17-й хромосомы (17p), и иногда его называют 17p-синдром.
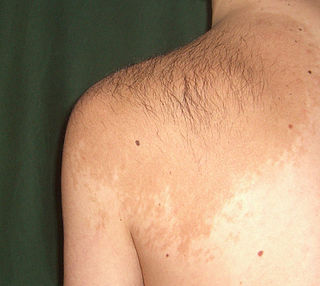
Невус Беккера, меланоз Беккера, пигментированный волосатый эпидермальный невус — один из гипертрихозов (дерматозов), доброкачественное новообразование кожи, появляющееся обычно на втором или третьем десятилетии жизни. У мужчин оно встречается в пять-шесть раз чаще, чем у женщин. По мере полового созревания образование склонно покрываться более толстыми, жесткими и темными волосами, чем остальное тело, поэтому может доставлять косметические неудобства. Участки кожи, поражённые невусом Беккера, с возрастом склонны темнеть, а изначально разрозненные островки могут сливаться в общий конгломерат.