Серотониновый синдром — редкая, однако потенциально смертельно опасная реакция организма на приём относительно больших доз лекарственных веществ, триптофана или наркотиков, повышающих серотонинергическую передачу. Может возникать в результате отравлений, приёма лекарств в больших количествах, неблагоприятной реакции на комбинацию принимаемых лекарств или наркотиков, а также из-за рекреационного использования определённых наркотических средств. Нередко возникает при применении двух и более препаратов — риск этого расстройства особенно высок при сочетании антидепрессантов группы СИОЗС и группы ИМАО, приводящем к особенно тяжёлым случаям серотонинового синдрома.
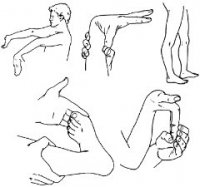
Синдром Э́лерса — Данло́са — это группа наследственных системных дисфункций соединительной ткани, вызванных дефектом в синтезе коллагена. В зависимости от отдельной мутации, серьёзность синдрома может измениться от умеренного до опасного для жизни. Лечения нет, но существует терапия (уход), смягчающая последствия.
5-я хромосо́ма челове́ка — одна из 23 пар человеческих хромосом. Хромосома содержит около 181 млн пар оснований, что составляет почти 6 % всего материала ДНК человеческой клетки. Являясь одной из самых больших человеческих хромосом, она тем не менее имеет одну из самых низких плотностей генов. Это частично объясняется наличием большого количества бедных генами участков, в которых наблюдается значительный уровень некодирующих консервативных последовательностей, идентичных имеющимся у немлекопитающих позвоночных, что позволяет предположить их функциональную важность. В настоящее время считается, что на 5-й хромосоме находятся от 900 до 1300 генов.
Синдром Торга-Винчестера — синдром, впервые описанный в 1969 году; у носителей которого отмечается спектр проявлений, в том числе сниженный рост вследствие патологии костей и суставов, помутнения роговицы, грубые черты лица, подкожные узелковые утолщения, огрубение кожи, гипертрихоз. Отклонения вызываются разнообразными мутациями гена MMP2. Их разнообразие привело к тому, что они были описаны как три различных синдрома: синдром Торга, синдром Винчестера, NAO-синдром, и лишь в 2006 году объединены под новым общим именем.

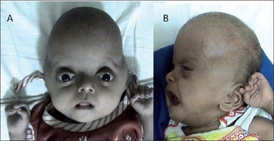
Синдром Робинова. Редкое генетическое заболевание, для которого на общем фоне отставания в развитии характерны — увеличенная голова, выпуклый лоб, низкий рост, полнота, брахидактилия конечностей, неразвитость внешних половых органов и др. Впервые синдром был описан в 1969 году в американском журнале заболеваний у детей педиатром и генетиком Мейнхардом Робиновым вместе с коллегами Фредериком Сильверманом и Хьюго Смитом. К 2002 году изучено и опубликовано в специализированной медицинской литературе более 100 клинических случаев заболевания. Синдром также известен как Робинов-Сильверман-Смит синдром. Рецессивная форма заболевания была ранее известна как синдром Covesdem.

Синдром Апе́ра (Аперта) — врожденная аномалия развития черепа, которая сочетается с отклонением развития кистей рук. Раннее закрытие венечного и стреловидного швов способствует деформации черепа, что приводит к внутричерепной гипертензии. Синдром Аперта является одной из форм акроцефалосиндактилии.

Синдро́м Да́уна — хромосомная болезнь, чаще всего вызванная тем, что хромосомы 21-й пары представлены тремя копиями вместо нормальных двух. Существует ещё две формы этого синдрома: транслокация хромосомы 21 на другие хромосомы — 4 % случаев, и мозаичный вариант синдрома — 5 %.

Синдро́м Шереше́вского — Тёрнера (СШТ) — хромосомный симптомокомплекс, обусловленный частичной или полной моносомией по X-хромосоме, в то время как здоровые женщины имеют и унаследованную от матери хромосому, и хромосому от отца. Характеризуется аномалиями физического развития, низкорослостью, половым инфантилизмом и другими разнящимися признаками. Традиционным методом верифицирования является кариотипирование. Терапия основана на приёме гормонов роста и эстрогенов.

Синдро́м Со́тоса — врождённое, в большинстве случаев спорадическое заболевание, впервые описанное в 60-х годах XX века. Характеризуется высокорослостью, крупным шишковатым черепом, выпуклым лбом, гипертелоризмом, наличием высокого нёба, антимонголоидным разрезом глаз, умеренной задержкой психического развития. Костный возраст опережает паспортный. Эндокринный статус нормальный.

Синдро́м Ларо́на — наследственное заболевание с аутосомно-рецессивным типом наследования — своеобразная разновидность карликовости (низкорослости), обусловленная врождённым дефектом гена рецептора соматотропного гормона (СТГ), приводящим к нечувствительности периферических тканей к действию гормона роста. Резистентность к ИФР-1 выявлена у африканских пигмеев.
Синдро́м де ля Шапе́ля — назван в честь исследователя, впервые охарактеризовавшего его в 1972 году. Синдром относится к редкой хромосомной патологии, возникающей в результате кроссинговера между X- и Y-хромосомами в процессе мейоза, в результате чего одна или обе X-хромосомы содержат нормальный мужской ген SRY. Распространённость данного синдрома составляет 4—5 на 100 000, что меньше встречаемости синдрома Клайнфельтера.
Синдро́м Гу́рлер — тяжёлое наследственное заболевание из группы мукополисахаридозов, относящихся к лизосомным болезням накопления. Характеризуется недостаточностью альфа-L-идуронидазы — фермента лизосом, участвующего в катаболизме кислых мукополисахаридов, которые составляют основу межклеточного вещества соединительной ткани.
Синдро́м Шейе — тяжёлое наследственное заболевание из группы мукополисахаридозов, относящихся к лизосомным болезням накопления. Характеризуется недостаточностью альфа-L-идуронидазы — фермента лизосом, участвующего в катаболизме кислых мукополисахаридов, которые составляют основу межклеточного вещества соединительной ткани. Заболевание редкое, проявляется в детском возрасте.

Мукополисахаридо́з I — группа метаболических заболеваний соединительной ткани, связанных с нарушением обмена кислых гликозаминогликанов, вызванных недостаточностью лизосомного фермента обмена гликозаминогликанов альфа-L-идуронидазы. Данная группа мукополисахаридозов связана с наследственной аномалией, вызванной генетически детерминированным дефектом гена, расположенного в локусе 4q16.3, которая наследуется по аутосомно-рецессивному типу. Проявляется в виде лизосомной болезни накопления, которая приводит к различным дефектам нервной, костной, хрящевой и соединительной тканей.
Синдром Коккейна , также называемый синдром Нил-Дингуолл — редкое аутосомно-рецессивное, нейродегенеративное расстройство, характеризующееся недостатком роста, нарушением развития нервной системы, аномальной чувствительностью к солнечному свету (фотосенсибилизация), заболеваниями глаз и преждевременным старением. Нездоровый вид и неврологические расстройства являются критериями для диагностики, а светочувствительность, нарушения слуха и ненормальные глаза — другие весьма общие черты. Возможны проблемы любого или всех внутренних органов. Это связано с группой расстройств, называемых лейкодистрофия. В основе расстройства лежит дефект механизма репарации ДНК. Интересно, в отличие от других дефектов репарации ДНК, пациенты с CS не предрасположены к раку или инфекции. Синдром Коккейна редок, но это разрушительная болезнь, которая, как правило, приводит к смерти в первом или втором десятилетии жизни. Мутация специфических генов в синдроме Коккейна известна, но широко распространенные эффекты и его отношения с репарацией ДНК еще не очень хорошо исследованы.

Рецептор фактора роста фибробластов 2 — мембранный белок, рецептор из семейства рецепторов фактора роста фибробластов. Продукт гена человека FGFR2. Как следует из названия белка, является рецептором факторов роста фибробластов. FGFR2 распознаёт кислый, основный факторы роста фибробластов и фактор роста кератиноцитов в зависимости от изоформы.
Синдром избытка ароматазы — редкий генный и эндокринный синдром, который характеризуется избыточной экспрессией ароматазы, фермента, ответственного за биосинтез эстрогенных половых гормонов из андрогенов, что, в свою очередь, приводит к чрезмерному уровню циркулирующих эстрогенов и, соответственно симптомы гиперэстрогенизма. Он затрагивает оба пола, проявляясь у мужчин как выраженная или полная фенотипическая феминизация за исключением гениталий, а у женщин - как гиперфеминизация.
Кардио-фацио-кожный синдром, КФКС, также известный как сердечно-кожно-лицевой синдром, кардио-фацио-кутанеальный синдром (КФКС), CFC Syndrome — редкое генетическое отклонение, Ras-патия, обусловленное мутациями в генах Ras / митоген-активированной протеинкиназы (MAPK): BRAF, MAP2K1, MAP2K2 и KRAS, которые, как правило, возникают de novo. Синдром характеризуется одновременным поражением сердечно-сосудистой системы, строения лица и кожных покровов, а также общей задержкой развития.
Синдром Смит — Магенис (СМС) — наследственное заболевание, имеет такие проявления, как умственная отсталость, аномалии лица, проблемы со сном и многочисленные поведенческие проблемы, в том числе и такие, как самоповреждение. Частота синдрома Смит — Магенис составляет 1 случай на 15 000-25 000 человек. Синдром Смит — Магенис, как правило, обусловлен микроделецией в коротком плече 17-й хромосомы (17p), и иногда его называют 17p-синдром.
Синдром Видемана — Раутенштрауха — чрезвычайно редкая врождённая патология, при которой человек преждевременно стареет. Больные этим синдромом практически не набирают вес, их кожа обвисает и выглядит старой, не соответствуя возрасту.