Боле́знь Ха́нтера — одна из форм мукополисахаридоза, мукополисахаридоз 2-го типа, редкое рецессивное Х‑сцепленное генетическое заболевание из группы лизосомных болезней накопления. Возникает в результате дефицита ряда ферментов, что приводит к накоплению белково‑углеводных комплексов и жиров в клетках.

Мукополисахаридо́зы сокращённо МПС, или англ. MPS — группа метаболических заболеваний соединительной ткани, связанных с нарушением обмена кислых гликозаминогликанов, связанных недостаточностью лизосомных ферментов обмена гликозаминогликанов. Заболевания вызваны наследственными аномалиями обмена, проявляются в виде лизосомной болезни накопления: различных дефектов костной, хрящевой, соединительной тканей.

Лизосо́мные боле́зни накопле́ния — общее название группы весьма редких наследственных заболеваний, вызванных нарушением функции внутриклеточных органелл лизосом. Эти одномембранные органоиды являются частью эндомембранной системы клетки и специализируются на внутриклеточном расщеплении веществ: гликогена, гликозаминогликанов, гликопротеинов и других. Лизосомные болезни накопления вызываются генетически обусловленным дефицитом ферментов лизосом, что приводит к накоплению макромолекул, являющихся субстратом этих ферментов, в различных органах и тканях организма.

Синдро́м Санфили́ппо — группа клинически сходных редких наследственных заболеваний, относящихся к лизосомным болезням накопления. Заболевания связаны с дефицитом того или иного фермента, в результате чего в лизосомах накапливается один тип гликозаминогликанов — гепарансульфат. Нарушения, в соответствии с дефектом гена, разделяют на четыре формы. Синдром описан в 1963 году американским учёным-педиатром Сильве́стром Санфили́ппо с соавторами. Пациенты с синдромом Санфилиппо обычно доживают лишь до юности или 1-го периода зрелости.

Боле́знь По́мпе — редкое, наследственное заболевание с аутосомно-рецессивным механизмом, наследования,, связанное с повреждением мышечных, и нервных клеток по всему организму. Клиническая картина данной патологии, обусловлена накоплением гликогена, в лизосомах, вызванным недостаточностью лизосомного фермента — кислой α-1,4-глюкозидазы. Различают быстро прогрессирующую (классическую) и медленно прогрессирующую формы болезни Помпе. Несмотря на широкий спектр клинических проявлений, гликогеноза II типа, в основе всех форм болезни лежит, дефицит одного фермента, кодируемого геном GAA.
Муколипидозы — собирательное название группы наследственных заболеваний, относящихся к лизосомным болезням накопления, связанных с дефицитом того или иного фермента. Гетерогенная группа болезней, объединяющая проявления недостаточности одного из ферментов лизосом, результатом которого является определённое сочетание накопления внутри клеток организма мукополисахаридов, гликопротеинов, олигосахаридов и гликолипидов. Изначально данная группа генных болезней, клиническая картина которых связана с нарушением нормального катаболизма различных субстратов внутри клеток, названа по аналогии с другими наследственными болезнями накопления. Открытие биохимических процессов, дефект которых приводил к развитию того или иного типа муколипидоза, внёс корректировку в классификацию. Изначально первые четыре типа были помечены как муколипидозы. Однако, теперь муколипидоз I типа (сиалидоз) классифицируется как гликопротеиноз, а муколипидоз IV типа (сиалолипидоз) — как ганглиозидоз.

Гаргоили́зм — характерная для болезни Пфаундлера — Гурлер и некоторых других типов мукополисахаридоза внешность: грубые черты лица: широкая и приплюснутая переносица, широко расставленные глаза, широкий с большими ноздрями нос, толстые губы и язык, открытый рот с маленькими редкими зубами; форма черепа нередко напоминает киль лодки. Заболевание обусловлено наследственной патологией соединительной ткани, проявляющейся поражением костей, суставов, глаз, внутренних органов и нервной системы. Понятие гаргоилизм берёт начало от фр. gargouille (гаргу́лья), обозначающего раструбы старинных водосточных труб средневековых соборов, украшавшиеся рельефными изображениями фантастических фигур с отталкивающим, причудливым лицом (химер).
Синдро́м Гу́рлер — тяжёлое наследственное заболевание из группы мукополисахаридозов, относящихся к лизосомным болезням накопления. Характеризуется недостаточностью альфа-L-идуронидазы — фермента лизосом, участвующего в катаболизме кислых мукополисахаридов, которые составляют основу межклеточного вещества соединительной ткани.

Метахромати́ческая лейкодистрофи́я — редкое наследственное заболевание из группы лизосомных болезней накопления с аутосомно-рецессивным механизмом наследования нарушения обмена веществ. Данная нозологическая единица из разряда лейкодистрофий относится к сфинголипидозам. Характеризуется недостаточностью арилсульфатазы А (цереброзидсульфатазы) — фермента лизосом, участвующего в метаболизме сфинголипидов, что вызывает накопление цереброзида сульфата.
Синдро́м Шейе — тяжёлое наследственное заболевание из группы мукополисахаридозов, относящихся к лизосомным болезням накопления. Характеризуется недостаточностью альфа-L-идуронидазы — фермента лизосом, участвующего в катаболизме кислых мукополисахаридов, которые составляют основу межклеточного вещества соединительной ткани. Заболевание редкое, проявляется в детском возрасте.
Синдро́м Гу́рлер — Шейе́ — наследственное заболевание из группы мукополисахаридозов, относящихся к лизосомным болезням накопления. Характеризуется недостаточностью альфа-L-идуронидазы — фермента лизосом, участвующего в катаболизме кислых мукополисахаридов, которые составляют основу межклеточного вещества соединительной ткани. Заболевание редкое, проявляется в детском возрасте.

Мукополисахаридо́з I — группа метаболических заболеваний соединительной ткани, связанных с нарушением обмена кислых гликозаминогликанов, вызванных недостаточностью лизосомного фермента обмена гликозаминогликанов альфа-L-идуронидазы. Данная группа мукополисахаридозов связана с наследственной аномалией, вызванной генетически детерминированным дефектом гена, расположенного в локусе 4q16.3, которая наследуется по аутосомно-рецессивному типу. Проявляется в виде лизосомной болезни накопления, которая приводит к различным дефектам нервной, костной, хрящевой и соединительной тканей.
Синдром Марото — Лами — редкая наследственная болезнь, одна из форм мукополисахаридоза из группы лизосомных болезней накопления, биохимически связанная с дефицитом фермента лизосом N-Ацетилгексозамин-4-сульфатсульфатазы.
GM2 ганглиозидо́з, вариа́нт АБ (англ. GM2-gangliosidosis, AB variant) — редкое аутосомно-рецессивное нарушение обмена веществ, связанное с мутацией гена GM2A. Характеризуется нормальной активностью β-гексозаминидаз А и Б и обусловлено недостаточностью активатора (белкового кофактора), необходимого для реализации ферментной активности в отношении субстрата. Заболевание клинически проявляется прогрессирующим разрушением нервных клеток головного и спинного мозга.
GM1-ганглиозидо́зы — редкие наследственные заболевания из группы лизосомных болезней накопления. Развитие клинической картины обусловлено дефектом или недостатком β-галактозидазы, который ведёт к нарушению метаболизма и накоплению субстратов (ганглиозида GM1, гликопротеинов и кератансульфата) главным образом в нервных клетках центральной и периферической нервной системы.

Синдром Сандхоффа — наследственное заболевание, вариант 0 GM2-ганглиозидоза из группы лизосомных болезней накопления — нарушение липидного обмена, вызванное наследственным дефицитом (недостаточностью) ферментов бета-гексозаминидазы А и Б.

Маннозидо́зы — редкие наследственные заболевания из группы лизосомных болезней накопления с аутосомно-рецессивным типом наследования.

Бета-маннозидоз — редкое аутосомно-рецессивное наследственное заболевание из группы лизосомных болезней накопления, связанное с нарушением метаболизма олигосахаридов в результате снижения активности фермента лизосом бета-маннозидазы.
Аспартилглюкозаминури́я — редкое аутосомно-рецессивное наследственное заболевание из группы лизосомных болезней накопления, связанное с нарушением метаболизма олигосахаридов в результате снижения активности фермента лизосом аспартилглюкозаминамидазы.
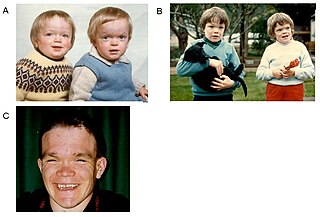
Альфа-маннозидо́з — редкое аутосомно-рецессивное наследственное заболевание из группы лизосомных болезней накопления, связанное с нарушением метаболизма олигосахаридов в результате снижения активности фермента лизосом альфа-маннозидазы. Также заболевание наносит ущерб в животноводстве.
