
Са́харный диабе́т 1-го ти́па — аутоиммунное заболевание эндокринной системы, основным диагностическим признаком которого является хроническая гипергликемия — повышенный уровень сахара в крови, полиурия, как следствие этого — жажда; потеря веса; чрезмерный либо сниженный аппетит; Сильное общее утомление организма; Боли в животе; при длительном проявлении болезни и отсутствии диагностики заболевания, начинается отравление организма продуктами распада жиров - часто проявляется в виде запаха ацетона от кожи, изо рта;

Целиаки́я, глютеновая энтеропатия — форма энтеропатии, генетически предрасположенная непереносимость глиадина или глютена. Это аутоиммунное заболевание с преимущественной локализацией в тонком кишечнике.
Синдром поликистозных яичников — полиэндокринный синдром, сопровождающийся нарушениями функции яичников, поджелудочной железы, коры надпочечников, гипоталамуса и гипофиза.
Синдром Ди Георга — разновидность идиопатического изолированного гипопаратиреоза; редкое врождённое заболевание. Генетической причиной синдрома Ди Георга является делеция центрального участка длинного плеча хромосомы 22 (22q11.2) размером 1.5-3 млн.п.н. Однако известны случаи, когда при тех же клинических проявлениях имеет место делеция других хромосом — 10р13, 17р13, 18q21 и других. В большинстве случаев делеция происходит во время мейоза при спермато- или овогенезе. Только в 5-10 % случаев дефектная хромосома наследуется по аутосомно-доминантному типу. Характеризуется агенезией или дисгенезом паращитовидных (околощитовидных) желёз, аплазией тимуса, приводящей к резкому снижению популяции Т-лимфоцитов и иммунологической недостаточности, врождёнными аномалиями крупных сосудов.
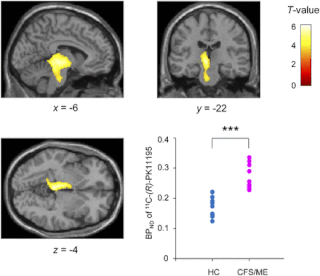
Синдро́м хрони́ческой уста́лости, миалги́ческий энцефаломиели́т (МЭ/СХУ, синдром утомляемости после перенесённой вирусной болезни, болезнь системной непереносимости нагрузок) — это тяжёлое хроническое системное заболевание, при котором нарушается работа иммунной, сердечно-сосудистой, нервной, гормональной и других систем организма.
Кахекси́я ; устар. худосо́чие — опасное для жизни патологическое состояние, крайнее истощение организма, которое характеризуется общей слабостью, резким снижением веса, активности физиологических процессов, а также изменением психического состояния больного, не старающегося активно похудеть. Кахексия физически ослабляет пациентов до состояния неподвижности, связанного с потерей аппетита, астенизацией и анемией, а реакция на стандартное лечение основного заболевания обычно плохая.

Синдро́м Ма́ртина — Белл — наследственное заболевание.
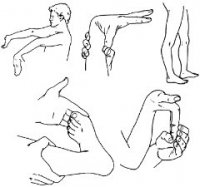
Синдром Э́лерса — Данло́са — это группа наследственных системных дисфункций соединительной ткани, вызванных дефектом в синтезе коллагена. В зависимости от отдельной мутации, серьёзность синдрома может измениться от умеренного до опасного для жизни. Лечения нет, но существует терапия (уход), смягчающая последствия.
Синдром Вискотта — Олдрича — редкое Х-сцепленное рецессивное заболевание, характеризующееся наличием экземы, тромбоцитопении, иммунодефицита, и кровавого поноса. Синоним — синдром экземы-тромбоцитопении-иммунодефицита в соответствии с оригинальным описанием Олдрича, сделанным в 1954 году.
Мно́жественная эндокри́нная неоплази́я (МЭН) — этот термин объединяет группу наследственных аутосомно-доминантных синдромов, обусловленных опухолями или гиперплазией нескольких эндокринных желез. Возможны также смешанные типы этих синдромов.

Синдро́м Шегре́на — аутоиммунное системное поражение соединительной ткани, проявляющееся вовлечением в патологический процесс желез внешней секреции, главным образом слюнных и слёзных, и хроническим прогрессирующим течением.
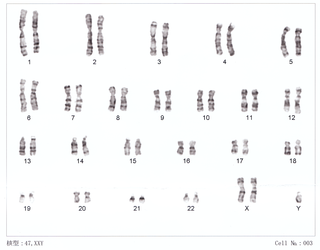
Синдром Клайнфельтера — генетическое отклонение. Клиническая картина синдрома описана в 1942 году в работах Гарри Клайнфельтера и Фуллера Олбрайта. Генетической особенностью этого синдрома является разнообразие цитогенетических вариантов и их сочетаний (мозаицизм). Обнаружено несколько типов полисомии по хромосомам X и Y у лиц мужского пола: 47, XXY; 47, XYY; 48, XXXY; 48, XYYY; 48 XXYY; 49 XXXXY; 49 XXXYY. Наиболее распространён синдром Клайнфельтера. Общая частота его колеблется в пределах 1 на 500—700 новорождённых мальчиков, что делает данный синдром первым по частоте встречаемости среди хромосомных болезней.

Синдро́м Шереше́вского — Тёрнера (СШТ) — хромосомный симптомокомплекс, обусловленный частичной или полной моносомией по X-хромосоме, в то время как здоровые женщины имеют и унаследованную от матери хромосому, и хромосому от отца. Характеризуется аномалиями физического развития, низкорослостью, половым инфантилизмом и другими разнящимися признаками. Традиционным методом верифицирования является кариотипирование. Терапия основана на приёме гормонов роста и эстрогенов.

Множественная эндокринная неоплазия типа IIb — в данный синдром вошли те же опухоли, что и в МЭН IIa, однако первичный гиперпаратиреоз проявляется реже, а медуллярная карцинома щитовидной железы протекает крайне злокачественно. Выявляется в возрасте 4—37 лет. Отличается от МЭН IIa наличием нейрином (невром) слизистых оболочек и расстройств опорно-двигательного аппарата. Нейриномы чаще локализуются на губах, щеках, языке, других отделах желудочно-кишечного тракта и представляют собой бело-розовые безболезненные узелки диаметром от 1—3 мм до 1 см. Множественная эндокринная неоплазия типа IIb является самым серьёзным вариантом синдрома МЭН.
Синдро́м MEDAC — характеризуется монилиазом слизистых оболочек и кожи, сахарным диабетом (редко), гипопаратиреозом и недостаточностью надпочечников. В 2/3 случаев синдром выявляется в неполной форме, включающей два из трёх основных компонентов.
Аутоимму́нный полиэндокри́нный синдро́м — аутоиммунное заболевание, объединяющее поражение нескольких эндокринных органов.
Синдром Макла — Уэльса (MWS) — это мутация в гене CIAS1 с развитием холодового аутовоспалительного синдрома. Является редким аутовоспалительным заболеванием наследственного характера. Преимущественно этническая распространённость — народы Северной Европы. Тип наследования — аутосомно-доминантный. Генетическая основа — мутация гена CIAS1, расположенного на длинном плече 1-й хромосомы (1q44) и кодирующего белка криопирина. Данный белок является основной образуемого в клетке супрамолекулярного комплекса, называемого инфламмасомой, выполняющего функцию превращения pro-IL-1β в активную форму, а также принимающего участие в выполнении программы апоптоза. Холодовой аутовоспалительный синдром тесно связан с двумя другими синдромами: семейной холодовой крапивницы и мультисистемным воспалительным заболеванием неонатального возраста — фактически, все связаны с мутациями гена. В целом Синдром Макла — Уэльса относится к группе криопирин-связанных периодических синдромов (CAPS).
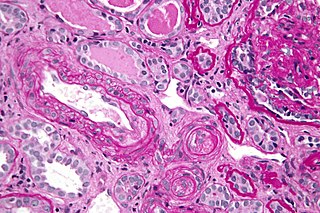
Антифосфолипидный синдром (АФС), или фосфолипидный синдром, или синдром антифосфолипидных антител — аутоиммунное состояние гиперкоагуляции, вызванное антифосфолипидными антителами. АФС провоцирует образование тромбов (тромбоз) как в артериях, так и венах, а также связанные с беременностью осложнения, такие как выкидыш, мертворождение, преждевременные роды и тяжелая преэклампсия.
Синдром 49, XXXXY — чрезвычайно редкая анеуплоидная половая хромосомная аномалия. Случается примерно в 1 из 85 000 до 100 000 случаев. Этот синдром является результатом материнского нерасхождения хромосом во время мейоза I и II. Впервые он был диагностирован в 1960 году и был назван синдромом Фраккаро по имени исследователя.
Аутоиммунитет – это система иммунных реакций организма на собственные здоровые клетки и ткани. Любое заболевание, которое является результатом такого аберрантного иммунного ответа, называют «аутоиммунным заболеванием». Яркими примерами являются целиакия, постинфекционный СРК, сахарный диабет 1-го типа, геморрагический васкулит, саркоидоз, системная красная волчанка (СКВ), синдром Шегрена, синдром CHARGE, аутоиммунный тиреоидит, диффузный токсический зоб, идиопатическая тромбоцитопеническая пурпура, болезнь Аддисона, ревматоидный артрит (РА), болезнь Бехтерева, полимиозит (ПМ), дерматомиозит (ДМ) и рассеянный склероз (РС). Аутоиммунные заболевания очень часто лечат стероидами.