
Альбинизм — наследственное заболевание, полное или почти полное отсутствие пигмента меланина или хлорофилла. Проявляется отсутствием нормальной вида окраски кожи, волос, шерсти, радужной и пигментной оболочек глаз, зелёных частей растений.

Синдром Ретта — психоневрологическое наследственное заболевание, встречается почти исключительно у девочек с частотой 1:10000 — 1:15000, является причиной тяжёлой умственной отсталости у девочек.

Синдро́м сава́нта, саванти́зм — редкое состояние, при котором люди с интеллектуальными или когнитивными нарушениями демонстрируют выдающиеся способности или таланты в определённых узких областях. Эти таланты могут проявляться в различных сферах, включая математику, музыку, искусство, память и другие. Хотя синдром саванта часто ассоциируется с расстройствами аутистического спектра (РАС), он также может встречаться у людей с нормальным или высоким уровнем интеллекта.

Синдром Ангельмана — обусловленная генетической аномалией патология, характеризующаяся такими признаками, как задержка психического развития, нарушения сна, припадки, хаотические движения, частый смех или улыбки. Также эту болезнь называют «синдром Петрушки» или «синдром счастливой куклы».

Синдром Вольфа — Хиршхорна — редкое генетическое заболевание, возникающее в результате делеции короткого плеча 4-й хромосомы. Впервые описан в 1965 году.

Синдро́м Ма́ртина — Белл — наследственное заболевание.

Синдро́м Альстрёма — генетическая патология человека, относящаяся к группе цилиопатий. Характеризуется пигментной дегенерацией сетчатки, ожирением, прогрессирующей нейросенсорной глухотой, дилятационной кардиомиопатией, сахарным диабетом и нефропатией. Впервые описан в 1959 году шведским психиатром Карлом-Генри Альстрёмом.

Синдром Э́двардса — хромосомное заболевание, характеризуется комплексом множественных пороков развития и трисомией 18 хромосомы. Описан в 1960 году Джоном Эдвардсом. Популяционная частота примерно 1:3000 в США и 1:5000 в мире на 2016 год. Дети с трисомией в 18-й хромосоме чаще рождаются у пожилых матерей, взаимосвязь с возрастом матери менее выражена, чем в случаях трисомии хромосомы 21 и 13. Для женщин старше 45 лет риск родить больного ребёнка составляет 0,7 %. Девочки с синдромом Эдвардса рождаются в три раза чаще мальчиков. Выживание после года жизни составляет около 5—10 %.

Синдро́м Да́уна — хромосомная болезнь, чаще всего вызванная тем, что хромосомы 21-й пары представлены тремя копиями вместо нормальных двух. Существует ещё две формы этого синдрома: транслокация хромосомы 21 на другие хромосомы — 4 % случаев, и мозаичный вариант синдрома — 5 %.
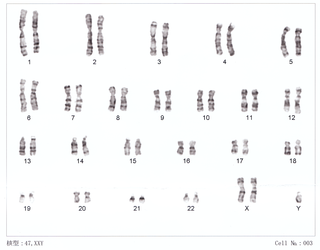
Синдром Клайнфельтера — генетическое отклонение. Клиническая картина синдрома описана в 1942 году в работах Гарри Клайнфельтера и Фуллера Олбрайта. Генетической особенностью этого синдрома является разнообразие цитогенетических вариантов и их сочетаний (мозаицизм). Обнаружено несколько типов полисомии по хромосомам X и Y у лиц мужского пола: 47, XXY; 47, XYY; 48, XXXY; 48, XYYY; 48 XXYY; 49 XXXXY; 49 XXXYY. Наиболее распространён синдром Клайнфельтера. Общая частота его колеблется в пределах 1 на 500—700 новорождённых мальчиков, что делает данный синдром первым по частоте встречаемости среди хромосомных болезней.

Синдро́м Шереше́вского — Тёрнера (СШТ) — хромосомный симптомокомплекс, обусловленный частичной или полной моносомией по X-хромосоме, в то время как здоровые женщины имеют и унаследованную от матери хромосому, и хромосому от отца. Характеризуется аномалиями физического развития, низкорослостью, половым инфантилизмом и другими разнящимися признаками. Традиционным методом верифицирования является кариотипирование. Терапия основана на приёме гормонов роста и эстрогенов.
Анеуплоиди́я — изменение кариотипа, при котором число хромосом в клетках не кратно гаплоидному набору (n). Отсутствие в хромосомном наборе диплоидного организма одной хромосомы называется моносомией (2n-1); отсутствие двух гомологичных хромосом — нуллисомией (2n-2); наличие дополнительной хромосомы называется трисомией (2n+1). Анеуплоидия возникает в результате нарушения сегрегации хромосом в митозе или мейозе. Анеуплоидия вызывает у человека некоторые наследственные синдромы. Анеуплоидия по аутосомам нарушает нормальное эмбриональное развитие и является одной из основных причин спонтанных абортов. Анеуплоидия характерна для опухолевых клеток, особенно для клеток сóлидных опухолей. Патологический фенотип при анеуплоидии формируется из-за нарушения дозового баланса генов, при моносомии дополнительный негативный вклад оказывает гемизиготное состояние генов моносомной хромосомы.
Синдро́м Пехкра́нца — Баби́нского — Фре́лиха — нейроэндокринный синдром, характеризующийся прогрессирующим ожирением с преимущественным отложением жира в плечевом поясе, молочных железах, на животе, ягодицах, бёдрах. Он сопровождается недоразвитием половых органов.
Синдро́м де ля Шапе́ля — назван в честь исследователя, впервые охарактеризовавшего его в 1972 году. Синдром относится к редкой хромосомной патологии, возникающей в результате кроссинговера между X- и Y-хромосомами в процессе мейоза, в результате чего одна или обе X-хромосомы содержат нормальный мужской ген SRY. Распространённость данного синдрома составляет 4—5 на 100 000, что меньше встречаемости синдрома Клайнфельтера.

Синдром Питта — Хопкинса — редкое генетическое заболевание, которое характеризуется умственной отсталостью, широким ртом и отличительными чертами лица, а также прерывистой гипервентиляцией с одышкой. По мере дальнейшего изучения синдрома Питта-Хопкинса спектр расстройств развития всё расширяется и может также включать проблемы с повышенной тревожностью, аутизмом, СДВГ и сенсорными расстройствами. Это связано с аномалией в хромосоме 18. В частности, это вызвано недостаточной экспрессией гена TCF4.

XYY-синдром, также известен как YY-синдром или Синдром Джейкобс, — хромосомное заболевание, характерное только для биологических мужчин. Носитель синдрома имеет дополнительную Y-хромосому, общий хромосомный набор составляет 44 аутосомы и три половые хромосомы. Внешне мужчины с дополнительной Y-хромосомой обычно не имеют существенных отличий от мужчин с обычным набором хромосом, но могут иметь ряд особенностей.
Синдром Коккейна , также называемый синдром Нил-Дингуолл — редкое аутосомно-рецессивное, нейродегенеративное расстройство, характеризующееся недостатком роста, нарушением развития нервной системы, аномальной чувствительностью к солнечному свету (фотосенсибилизация), заболеваниями глаз и преждевременным старением. Нездоровый вид и неврологические расстройства являются критериями для диагностики, а светочувствительность, нарушения слуха и ненормальные глаза — другие весьма общие черты. Возможны проблемы любого или всех внутренних органов. Это связано с группой расстройств, называемых лейкодистрофия. В основе расстройства лежит дефект механизма репарации ДНК. Интересно, в отличие от других дефектов репарации ДНК, пациенты с CS не предрасположены к раку или инфекции. Синдром Коккейна редок, но это разрушительная болезнь, которая, как правило, приводит к смерти в первом или втором десятилетии жизни. Мутация специфических генов в синдроме Коккейна известна, но широко распространенные эффекты и его отношения с репарацией ДНК еще не очень хорошо исследованы.
Синдром 49, XXXXY — чрезвычайно редкая анеуплоидная половая хромосомная аномалия. Случается примерно в 1 из 85 000 до 100 000 случаев. Этот синдром является результатом материнского нерасхождения хромосом во время мейоза I и II. Впервые он был диагностирован в 1960 году и был назван синдромом Фраккаро по имени исследователя.

Синдром 48, XXYY — это аномалия хромосом, при которой у человека имеется дополнительная X и Y-хромосома. Клетки человека обычно содержат две половые хромосомы, одну от матери и одну от отца. Обычно женщины имеют две Х-хромосомы (XX), а мужчины имеют одну Х и одну Y-хромосому (XY). Появление по крайней мере одной Y-хромосомы с правильно функционирующим геном SRY делает человека физиологически мужчиной. Следовательно, люди с XXYY являются генотипически мужчинами. Мужчины с синдромом XXYY имеют 48 хромосом вместо типичных 46. По оценкам, он случается в 1 случае на каждые 18 000-40 000 новорожденных.
Синдром 48, XXXY — генетическое состояние, обусловленное анеуплоидией половых хромосом, когда у человека есть две дополнительные хромосомы. Обычно мужчины имеют только две половые хромосомы, X и Y, а женщины две X-хромосомы. Присутствие одной Y-хромосомы с функционирующим геном SRY вызывает экспрессию генов, определяющих мужской пол. Из-за этого синдром XXXY затрагивает только мужчин с XY. Существует широкий спектр симптомов, связанных с этим синдромом, включая когнитивные и поведенческие проблемы, тауродонтизм и бесплодие. Этот синдром обычно возникает через новую мутацию в одной из гамет родителей, поскольку люди с этим кариотипом обычно бесплодны. По оценкам, XXXY встречается в 1 случае из 50 000 рождённых мужчин.
