Синдром Экарди — Гутьер
| Синдром Экарди — Гутьер | |
|---|---|
| МКБ-10 | G93.4 |
| МКБ-10-КМ | G31.8 |
| МКБ-9 | 277.2 |
| OMIM | 225750, 610181, 610329, 610333, 612952, 615010, 615846 и 225750 |
| DiseasesDB | 31680 |
| MeSH | C535607 |
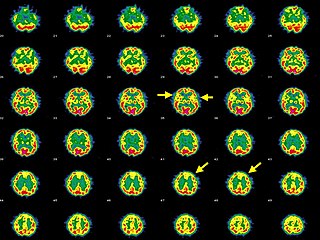
Синдром Экарди — Гутьер (также синдром Айкарди — Гутьереса, AGS (от англ., Aicardi–Goutières syndrome)) — является редким, обычно начинающимся в раннем детстве, воспалительным заболеванием, чаще всего поражающим мозг и кожу[1][2]. У большинства пациентов наблюдаются серьёзные соматические проявления и умственная отсталость. Клинические признаки синдрома могут имитировать признаки внутриутробной инфекции, а некоторые симптомы также совпадают с системной красной волчанкой[3][4][5]. Первое описание восьми случаев синдрома было сделано в 1984 г.[1], болезнь получила название «синдром Экарди-Гутьер» в 1992 году[6], первая конференция, посвященная этому заболеванию, была проведена в Павии в 2001 году[7].
Этиология
Синдром Экарди-Гутьер является генетически гетерогенным заболеванием, с заболеванием связаны мутации в семи генах, а именно: TREX1[8], RNASEH2A, RNASEH2B, RNASEH2C (которые вместе кодируют ферментный комплекс рибонуклеазы H2)[9], SAMHD1[10], ADAR1[11] и IFIH1[12]. Это неврологическое заболевание встречается у всех групп населения во всем мире. На 2014 год было известно не менее 400 случаев синдрома[13].
| Тип | OMIM | Ген | Локус | Частота |
|---|---|---|---|---|
| AGS1 | OMIM 225750 | TREX1 | 3p21.31 | 23% (1% с доминантным наследованием) |
| AGS2 | OMIM 610181 | RNASEH2B | 13q14.3 | 36% |
| AGS3 | OMIM 610329 | RNASEH2C | 11q13.1 | 12% |
| AGS4 | OMIM 610333 | RNASEH2A | 19p13.2 | 5% |
| AGS5 | OMIM 612952 | SAMHD1 | 20q11.23 | 13% |
| AGS6 | OMIM 615010 | ADAR | 1q21.3 | 7% (1% с доминантным наследованием) |
| AGS7 | OMIM 615846 | IFIH1 | 2q24 | 3% (все с доминантным наследованием) |
Примечания
- ↑ 1 2 Aicardi J, Goutieres F (1984). "A progressive familial encephalopathy in infancy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis". Ann Neurol. 15 (1): 49—54. doi:10.1002/ana.410150109. PMID 6712192.
- ↑ Tolmie JL; Shillito P; Hughes-Benzie R; Stephenson JB (1995). "The Aicardi-Goutieres syndrome (familial, early onset encephalopathy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis)". J Med Genet. 32 (11): 881—884. doi:10.1136/jmg.32.11.881. PMC 1051740. PMID 8592332.
- ↑ Aicardi, J; Goutieres, F (2000). "Systemic lupus erythematosus or Aicardi-Goutieres syndrome?". Neuropediatrics. 31 (3): 113. doi:10.1055/s-2000-7533. PMID 10963096.
- ↑ Dale RC; Tang SP; Heckmatt JZ; Tatnall FM (2000). "Familial systemic lupus erythematosus and congenital infection-like syndrome". Neuropediatrics. 31 (3): 155—158. doi:10.1055/s-2000-7492. PMID 10963105.
- ↑ Crow, YJ; Livingston, JH (2008). "Aicardi-Goutieres syndrome: an important Mendelian mimic of congenital infection". Dev Med Child Neurol. 50 (6): 410—416. doi:10.1111/j.1469-8749.2008.02062.x. PMID 18422679.
- ↑ Bonnemann, CG; Meinecke, P (1992). "Encephalopathy of infancy with intracerebral calcification and chronic spinal fluid lymphocytosis - another case of the Aicardi-Goutieres syndrome". Neuropaediatrics. 23 (3): 157—61. doi:10.1055/s-2008-1071333. PMID 1641084.
- ↑ "Proceedings of the International Meeting on Aicardi-Goutieres Syndrome Pavia, Italy, 28-29 May 2001". Eur J Paediatr Neurol. 6, Suppl A: A1—86. 2002.
- ↑ Crow, YJ; et al. (2006). "Mutations in the gene encoding the 3'-5' DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus". Nat Genet. 38 (8): 917—20. doi:10.1038/ng1845. PMID 16845398.
- ↑ Crow, YJ; et al. (2006). "Mutations in the genes encoding ribonuclease H2 subunits cause Aicardi-Goutieres syndrome and mimic congenital viral brain infection". Nat Genet. 38 (8): 910—6. doi:10.1038/ng1842. PMID 16845400.
- ↑ Rice, GI; et al. (2009). "Mutations involved in Aicardi-Goutieres syndrome implicate SAMHD1 as regulator of the innate immune response". Nat Genet. 41 (7): 829—32. doi:10.1038/ng.373. PMC 4154505. PMID 19525956.
- ↑ Rice, GI; et al. (2012). "Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type 1 interferon signature". Nat Genet. 44 (11): 1243—8. doi:10.1038/ng.2414. PMC 4154508. PMID 23001123.
- ↑ Rice, GI; et al. (2014). "Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type 1 interferon signaling". Nat Genet. 46 (5): 503—509. doi:10.1038/ng.2933. PMC 4004585. PMID 24686847.
- ↑ Брюханова Н. О. и др. Синдром Айкарди-Гутьерес у детей с идиопатической эпилепсией // Российский вестник перинатологии и педиатрии. — 2016. — Т. 61, № 2. — С. 68-75. — doi:10.21508/1027-4065-2016-61-2-68-75. Архивировано 21 января 2021 года.
Ссылки
- Айкарди-Гутьереса синдром
- Корк А., Петров С. Один из пятисот. Как живет мальчик с синдромом Айкарди — Гутьерес. https://www.pravmir.ru. Правмир (29 июля 2020). Дата обращения: 17 января 2021.