Ше́рер — немецкая фамилия. Многие французы с фамилией Шерер ведут происхождение из Эльзаса, неоднократно менявшего принадлежность между Францией и Германией.

Мукополисахаридо́зы сокращённо МПС, или англ. MPS — группа метаболических заболеваний соединительной ткани, связанных с нарушением обмена кислых гликозаминогликанов, связанных недостаточностью лизосомных ферментов обмена гликозаминогликанов. Заболевания вызваны наследственными аномалиями обмена, проявляются в виде лизосомной болезни накопления: различных дефектов костной, хрящевой, соединительной тканей.

Синдро́м Санфили́ппо — группа клинически сходных редких наследственных заболеваний, относящихся к лизосомным болезням накопления. Заболевания связаны с дефицитом того или иного фермента, в результате чего в лизосомах накапливается один тип гликозаминогликанов — гепарансульфат. Нарушения, в соответствии с дефектом гена, разделяют на четыре формы. Синдром описан в 1963 году американским учёным-педиатром Сильве́стром Санфили́ппо с соавторами. Пациенты с синдромом Санфилиппо обычно доживают лишь до юности или 1-го периода зрелости.
Синдро́м Гу́рлер — тяжёлое наследственное заболевание из группы мукополисахаридозов, относящихся к лизосомным болезням накопления. Характеризуется недостаточностью альфа-L-идуронидазы — фермента лизосом, участвующего в катаболизме кислых мукополисахаридов, которые составляют основу межклеточного вещества соединительной ткани.
Боле́знь Бильшо́вского — Янско́го — поздняя инфантильная (детская) форма восковидного липофусциноза нейронов, которая развивается на фоне дефицита лизосомного фермента трипептидил-пептидазы-1. Относится к группе лизосомных болезней накопления.
Синдро́м Шейе — тяжёлое наследственное заболевание из группы мукополисахаридозов, относящихся к лизосомным болезням накопления. Характеризуется недостаточностью альфа-L-идуронидазы — фермента лизосом, участвующего в катаболизме кислых мукополисахаридов, которые составляют основу межклеточного вещества соединительной ткани. Заболевание редкое, проявляется в детском возрасте.
Синдром Марото — Лами — редкая наследственная болезнь, одна из форм мукополисахаридоза из группы лизосомных болезней накопления, биохимически связанная с дефицитом фермента лизосом N-Ацетилгексозамин-4-сульфатсульфатазы.

GM2-ганглиозидо́з — тяжёлое наследственное заболевание, развивающееся в результате дефицита или недостаточной активности фермента гексозаминидазы и накопления в клетках ганглиозидов. Относится к лизосомным болезням накопления и имеет три варианта, связанные с мутациями в разных генах, которые оказывают вляние на активность общей гексозаминидазы. Два варианта заболевания больше известны под индивидуальными именами, полученными в честь авторов, впервые описавших их клиническую картину: болезнь Тея — Сакса, болезнь Сандхоффа. Третий вариант этой болезни носит название GM2 ганглиозидоз, вариант АБ. Болезнь Тея — Сакса вызвана мутацией в гене HEXA, кодирующий альфа-субъединицу гексоаминидазы А. Болезнь Сандхоффа вызвана мутацией в гене HEXB, кодирующий бета-субъединицу гексоаминидаз А и Б. GM2 ганглиозидоз, АБ вариант связан с нарушением в гене GM2, кодирующий белок-активатор GM2A.
GM1-ганглиозидо́зы — редкие наследственные заболевания из группы лизосомных болезней накопления. Развитие клинической картины обусловлено дефектом или недостатком β-галактозидазы, который ведёт к нарушению метаболизма и накоплению субстратов (ганглиозида GM1, гликопротеинов и кератансульфата) главным образом в нервных клетках центральной и периферической нервной системы.

Синдром Сандхоффа — наследственное заболевание, вариант 0 GM2-ганглиозидоза из группы лизосомных болезней накопления — нарушение липидного обмена, вызванное наследственным дефицитом (недостаточностью) ферментов бета-гексозаминидазы А и Б.
Синдро́м Сла́я — редкое наследственное заболевание из группы мукополисахаридозов, относящееся к лизосомным болезням накопления, характеризуется дефицитом фермента лизосом β-глюкуронидазы. В свою очередь, дефицит β-глюкуронидазы ведёт к накоплению определённых сложных углеводов (мукополисахаридов) во многих тканях и органах тела человека.
Болезнь Вольмана — – это редкое аутосомно-рецессивное заболевание лизосомального накопления, вызванное повреждающими мутациями гена LIPA. Возраст начала заболевания и темпы его прогрессирования в значительной степени вариабельны и могут быть связаны с природой, лежащих в основе мутаций. Заболевание у пациентов грудного возраста имеет наиболее быстро прогрессирующее течение с развитием проявлений и симптомов в первые недели жизни; эти пациенты редко доживают до возраста, превышающего 6 месяцев. У детей старшего возраста и взрослых заболевание, обычно, характеризуется определенным сочетанием дислипидемии, гепатомегалии, повышением уровня трансаминаз и микровезикулярным стеатозом в биопсийном материале. У большей части пациентов наблюдается повреждение печени с исходом в фиброз, цирроз и печеночную недостаточность. Частыми изменениями являются повышение уровней холестерина липопротеинов низкой плотности и снижение уровней холестерина липопротеинов высокой плотности. Начиная с детского возраста, могут проявляться и нарушения со стороны сердечно-сосудистой системы. Учитывая, что эти клинические проявления могут наблюдаться и при других сердечно-сосудистых нарушениях, заболеваниях печени и метаболических расстройствах, неудивительно, что LAL-D часто не диагностируется в клинической практике.
Псевдополидистрофи́я Гу́рлер — наследственное заболевание из группы муколипидозов, относящееся к лизосомным болезням накопления с аутосомно-рецессивным механизмом наследования нарушения обмена веществ. Клинически представляет собой заболевание с фенотипическими признаками мукополисахаридоза, напоминает более лёгкий вариант I-клеточной болезни. Данный симптомокомплекс получил название псевдо-Гурлер в связи с клинической схожестью симптомов с одним из представителей мукополисахаридоза — синдромом Гурлер.
Недоста́точность ки́слой фосфата́зы — редкое наследственное заболевание из группы лизосомных болезней накопления. Клиническая симптоматика дефицита кислой фосфатазы развивается в раннем возрасте. Характеризуется гепатомегалией и задержкой психического развития.

Маннозидо́зы — редкие наследственные заболевания из группы лизосомных болезней накопления с аутосомно-рецессивным типом наследования.
Боле́знь Ку́фса — редкое наследственное нейродегенеративное заболевание с летальным исходом из группы лизосомных болезней накопления, которое развивается на фоне дефицита фермента лизосом и наследуется по аутосомно-рецессивному типу.

Аутосо́мно-рецесси́вное насле́дование — свойственный диплоидным эукариотам тип наследования признака, контролируемого рецессивными аллелями аутосомного гена. Для проявления мутации или болезни с таким типом наследования мутантный аллель, локализованный в аутосоме, должен быть унаследован от обоих родителей. Иными словами, мутация проявляется только в гомозиготном состоянии, то есть тогда, когда обе копии гена, расположенные на гомологичных аутосомах, являются повреждёнными. Если мутация находится в гетерозиготном состоянии, и мутантному аллелю сопутствует нормальный функциональный аллель, то аутосомно-рецессивная мутация не проявляется.

Бета-маннозидоз — редкое аутосомно-рецессивное наследственное заболевание из группы лизосомных болезней накопления, связанное с нарушением метаболизма олигосахаридов в результате снижения активности фермента лизосом бета-маннозидазы.
Аспартилглюкозаминури́я — редкое аутосомно-рецессивное наследственное заболевание из группы лизосомных болезней накопления, связанное с нарушением метаболизма олигосахаридов в результате снижения активности фермента лизосом аспартилглюкозаминамидазы.
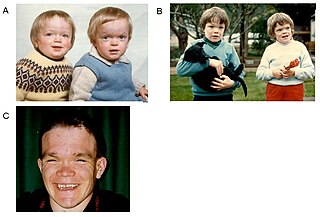
Альфа-маннозидо́з — редкое аутосомно-рецессивное наследственное заболевание из группы лизосомных болезней накопления, связанное с нарушением метаболизма олигосахаридов в результате снижения активности фермента лизосом альфа-маннозидазы. Также заболевание наносит ущерб в животноводстве.

