
Рецептор липопротеинов очень низкой плотности по структуре схож с рецептором липопротеинов низкой плотности, но не способен связывать липопротеины низкой плотности. Рецептор VLDLR играет роль в метаболизме липопротеинов очень низкой плотности. Экспрессия VLDLR высока в сердце, скелетных мышцах, жировой ткани; VLDLR совместно с рецептором LDLR связывает и захватывает остаточные липопротеины: липопротеины промежуточной плотности и хиломикронные ремнанты.

Синдро́м Праде́ра — Ви́лли — редкое наследственное заболевание, причиной которого является отсутствие отцовской копии участка хромосомы 15q11-13. В этом участке хромосомы 15 находятся гены, в регуляции которых задействован геномный импринтинг. Большинство случаев являются спорадическими, для редких описанных семейных случаев характерно неменделевское наследование.

Синдро́м Ма́ртина — Белл — наследственное заболевание.
5-я хромосо́ма челове́ка — одна из 23 пар человеческих хромосом. Хромосома содержит около 181 млн пар оснований, что составляет почти 6 % всего материала ДНК человеческой клетки. Являясь одной из самых больших человеческих хромосом, она тем не менее имеет одну из самых низких плотностей генов. Это частично объясняется наличием большого количества бедных генами участков, в которых наблюдается значительный уровень некодирующих консервативных последовательностей, идентичных имеющимся у немлекопитающих позвоночных, что позволяет предположить их функциональную важность. В настоящее время считается, что на 5-й хромосоме находятся от 900 до 1300 генов.
Цилиопатии — генетически обусловленные заболевания, возникающие при нарушении структуры или функции цилий.
Синдром Вискотта — Олдрича — редкое Х-сцепленное рецессивное заболевание, характеризующееся наличием экземы, тромбоцитопении, иммунодефицита, и кровавого поноса. Синоним — синдром экземы-тромбоцитопении-иммунодефицита в соответствии с оригинальным описанием Олдрича, сделанным в 1954 году.

Синдром Апе́ра (Аперта) — врожденная аномалия развития черепа, которая сочетается с отклонением развития кистей рук. Раннее закрытие венечного и стреловидного швов способствует деформации черепа, что приводит к внутричерепной гипертензии. Синдром Аперта является одной из форм акроцефалосиндактилии.
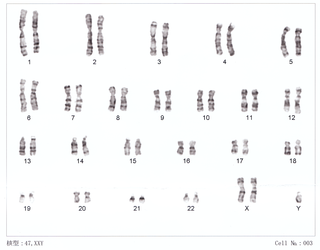
Синдром Клайнфельтера — генетическое отклонение. Клиническая картина синдрома описана в 1942 году в работах Гарри Клайнфельтера и Фуллера Олбрайта. Генетической особенностью этого синдрома является разнообразие цитогенетических вариантов и их сочетаний (мозаицизм). Обнаружено несколько типов полисомии по хромосомам X и Y у лиц мужского пола: 47, XXY; 47, XYY; 48, XXXY; 48, XYYY; 48 XXYY; 49 XXXXY; 49 XXXYY. Наиболее распространён синдром Клайнфельтера. Общая частота его колеблется в пределах 1 на 500—700 новорождённых мальчиков, что делает данный синдром первым по частоте встречаемости среди хромосомных болезней.

DIDMOAD-синдро́м — аутосомно-рецессивно наследуемый синдром, ассоциированный с инсулинозависимым сахарным диабетом и прогрессирующей атрофией диска зрительного нерва, которые выявляют до 16-летнего возраста. Сочетается с двусторонней прогрессирующей нейросенсорной тугоухостью, несахарным диабетом центрального генеза, дисфункцией автономной нервной системы, приводящей к развитию нейропатического мочевого пузыря и другим проявлениям нейродегенерации, включающими мозжечковую атаксию, миоклональную эпилепсию и атрофию ствола головного мозга. Развёрнутая клиническая картина (фенотипически) встречается приблизительно у 75 % пациентов. Сахарный диабет неаутоиммунного генеза, клинические проявления недостаточности инсулина проявляются приблизительно в 6-летнем возрасте. Средняя продолжительность жизни достигает 30 лет, в течение этого срока происходит развитие полного фенотипа данного синдрома.

Синдром Рубинштейна-Тейби впервые описан в 1963 г. J. Rubinstein и Н. Taybi.
Популяционная частота — 1:25 000 — 30000.
Соотношение полов — M1:Ж1.
Тип наследования неизвестен.
PTEN — фосфатаза с двойной субстратной специфичностью, продукт гена PTEN. Субстратами этой фосфатазы могут быть как белки, так и фосфатидилинозитол-3-фосфаты. PTEN катализирует отщепление фосфатной группы в положении 3D инозитольного кольца фосфатидилинозитол-3-фосфатов, лишая их таким образом функций вторичных посредников при передаче сигнала в клетке. Эта фосфатаза является одним из немногих негативных регуляторов PI3K/AKT/mTOR-сигнального пути, что делает её антионкобелком. Ген PTEN часто бывает мутирован при различных типах злокачественных опухолей.

Синдром Блума или Синдром Блум-Торре-Мачэйкик — редкое аутосомно-рецессивное заболевание, для которого характерны низкий рост больных и предрасположенность к онкологическим заболеваниям. Клетки больных синдромом Блума демонстрируют сильную геномную нестабильность, в частности, частую гомологичную рекомбинацию. Заболевание было обнаружено и впервые описано американским дерматологом доктором Дэвидом Блумом в 1954 году.
RAI1 — белок, кодируемый одноименным геном, расположенным у людей на коротком плече 17-й хромосомы. Длина полипептидной цепи белка составляет 1906 аминокислот, а молекулярная масса — 203 352. Является фактором транскрипции, связанным с синдромом Смит-Магенис, когда у людей есть делеции этого гена или мутация в нём, и с синдромом Потоцкой-Лупского, когда в геноме наблюдается дупликация этого гена.

Белок XRP2 является белком, который у человека кодируется геном RP2.
Регулирующий интерферон фактор 6, известный также под названием IRF6, — белок, который у человека кодируется геном IRF6.
CHD7 известный также как АТФ-зависимая хеликаза CHD7 — фермент, который у человека кодируется геном CHD7.

Аладин (англ. Aladin), также известный как адракалин — белок, кодируемый у человека геном AAAS (англ. Aladin).
Синдром лёгкой нечувствительности к андрогенам (СЛНкА) — состояние, которое приводит к лёгкому ухудшению способности клеток реагировать на андрогены. Степень нарушения достаточна для нарушения сперматогенеза и / или развития вторичных половых признаков в период полового созревания у мужчин, но не влияет на дифференцировку или развитие гениталий. Нечувствительность к андрогенам существенно не влияет на развитие женских половых органов и половое развитие как таковой; СЛНкА диагностируется только у мужчин. Клинический фенотип, связанный с СЛНкА, представляет собой нормальную мужскую особь с лёгким дефектом сперматогенеза и / или уменьшенным количеством волос на теле.
Синдром Смит — Магенис (СМС) — наследственное заболевание, имеет такие проявления, как умственная отсталость, аномалии лица, проблемы со сном и многочисленные поведенческие проблемы, в том числе и такие, как самоповреждение. Частота синдрома Смит — Магенис составляет 1 случай на 15 000-25 000 человек. Синдром Смит — Магенис, как правило, обусловлен микроделецией в коротком плече 17-й хромосомы (17p), и иногда его называют 17p-синдром.
Синдром Экарди — Гутьер — является редким, обычно начинающимся в раннем детстве, воспалительным заболеванием, чаще всего поражающим мозг и кожу. У большинства пациентов наблюдаются серьёзные соматические проявления и умственная отсталость. Клинические признаки синдрома могут имитировать признаки внутриутробной инфекции, а некоторые симптомы также совпадают с системной красной волчанкой. Первое описание восьми случаев синдрома было сделано в 1984 г., болезнь получила название «синдром Экарди-Гутьер» в 1992 году, первая конференция, посвященная этому заболеванию, была проведена в Павии в 2001 году.

