
Насле́дственные заболева́ния — заболевания, возникновение и развитие которых связано с различными дефектами и нарушениями в наследственном аппарате клеток. В основе наследственных заболеваний лежат мутации: хромосомные, генные и митохондриальные. Наследственные заболевания могут быть обусловлены мутациями, передаваемыми в семьях по наследству, или мутациями, вновь возникшими в клетках зародышевой линии, в зиготе или на очень ранних этапах развития. Наследственные болезни многочисленны и разнообразны по проявлениям.

Синдром Холт — Орама, синдром сердце-рука — наследственное генетическое заболевание, поражающее развитие верхних конечностей и сердца. Обусловлен дефектом гена TBX5. Частота встречаемости — 1:100 000, 60 % случаев семейные, самцы и самки одинаково подвержены синдрому.
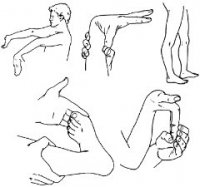
Синдром Э́лерса — Данло́са — это группа наследственных системных дисфункций соединительной ткани, вызванных дефектом в синтезе коллагена. В зависимости от отдельной мутации, серьёзность синдрома может измениться от умеренного до опасного для жизни. Лечения нет, но существует терапия (уход), смягчающая последствия.
Синдром Лея или подострая некротизирующая энцефаломиопатия — редкий наследственный нейрометаболический синдром, поражающий ЦНС. На сегодняшний день лечения данного заболевания не известно. У синдрома множество причин, значительную роль играют мутации генов, связанных с работой митохондрий. В сентябре 2016 года появилось сообщение о первой профилактике заболевания путём рождения ребёнка «от трёх родителей» — с использованием митохондрий донорской яйцеклетки.

Синдром Робинова. Редкое генетическое заболевание, для которого на общем фоне отставания в развитии характерны — увеличенная голова, выпуклый лоб, низкий рост, полнота, брахидактилия конечностей, неразвитость внешних половых органов и др. Впервые синдром был описан в 1969 году в американском журнале заболеваний у детей педиатром и генетиком Мейнхардом Робиновым вместе с коллегами Фредериком Сильверманом и Хьюго Смитом. К 2002 году изучено и опубликовано в специализированной медицинской литературе более 100 клинических случаев заболевания. Синдром также известен как Робинов-Сильверман-Смит синдром. Рецессивная форма заболевания была ранее известна как синдром Covesdem.

Синдром Пата́у — хромосомное заболевание человека, которое характеризуется наличием в клетках дополнительной хромосомы 13.
Синдром Свайера, XY дисгенезия гонад, женская гонадальная дисгенезия или гонадальная дисгенезия — генетическое нарушение, вариант гипогонадизма с кариотипом 46,XY. Организм человека с синдромом Свайера имеет характерный для мужского организма набор хромосом, но половые железы представляют собой гонадный тяж и не производят гормоны. В результате он имеет женские гениталии, женскую репродуктивную систему и выглядит как женщина. В период полового созревания развитие вторичных половых признаков не происходит и наблюдается аменорея. Существует практика удаления гонад в раннем возрасте с целью предотвращения развития рака.

Синдро́м Нуна́н — редкая врождённая патология, как правило, наследуется по аутосомно-доминантному типу, носит семейный характер, однако встречаются и спорадически. Синдром предполагает наличие фенотипа, характерного для синдрома Шерешевского-Тернера у особей женского и мужского пола с нормальным генотипом.
Синдро́м де ля Шапе́ля — назван в честь исследователя, впервые охарактеризовавшего его в 1972 году. Синдром относится к редкой хромосомной патологии, возникающей в результате кроссинговера между X- и Y-хромосомами в процессе мейоза, в результате чего одна или обе X-хромосомы содержат нормальный мужской ген SRY. Распространённость данного синдрома составляет 4—5 на 100 000, что меньше встречаемости синдрома Клайнфельтера.

Синдром Питта — Хопкинса — редкое генетическое заболевание, которое характеризуется умственной отсталостью, широким ртом и отличительными чертами лица, а также прерывистой гипервентиляцией с одышкой. По мере дальнейшего изучения синдрома Питта-Хопкинса спектр расстройств развития всё расширяется и может также включать проблемы с повышенной тревожностью, аутизмом, СДВГ и сенсорными расстройствами. Это связано с аномалией в хромосоме 18. В частности, это вызвано недостаточной экспрессией гена TCF4.
Пренатальная патология изучает все патологические процессы, возникающие в пренатальном периоде, а также различные нарушения созревания гамет.

Синдром Корнелии де Ланге — наследственное заболевание, проявляющееся умственной отсталостью и множественными аномалиями развития. Частота заболевания — примерно 1 на 10000.

X-сцепленное рецессивное наследование — один из видов сцепленного с полом наследования. Такое наследование характерно для признаков, гены которых расположены на Х-хромосоме и которые проявляются только в гомозиготном или гемизиготном состоянии. Такой тип наследования имеет ряд врождённых наследственных заболеваний у человека, эти заболевания связаны с дефектом какого-либо из генов, расположенных на половой Х-хромосоме, и проявляются в случае, если нет другой Х-хромосомы с нормальной копией того же гена. В литературе встречается сокращение XR для обозначения X-сцепленного рецессивного наследования.
Синдром Неймегена является редким аутосомно-рецессивным наследственным синдромом хромосомной нестабильности, при котором наблюдается микроцефалия, иммунодефицит и склонность к злокачественным новообразованиям. Синдром Неймегена вызван мутациями в гене NBN, который участвует в клеточном ответе на повреждения ДНК.
Трихотиодистрофия (TTD) — аутосомно-рецессивное наследственное заболевание, характеризующееся ломким волосом и интеллектуальными нарушениями. TTD связана с диапазоном симптомов, затрагивающих органы эктодермы и нейроэктодермы. TTD могут быть подразделены на четыре синдрома: Примерно половина всех пациентов с трихотиодистрофией страдают светочувствительностью, которая делит классификацию синдромов с ней или без неё; BIDS и PBIDS, IBIDS и PIBIDS. Современное использование включает TTD-P (светочувствительная) и TTD.

Тройной синдром (ААА), также известный как синдром Ахалазия-Аддисонианизма-Алакримии или синдром Оллгрова , — редкий аутосомно-рецессивный врожденный порок. В большинстве случаев характеризуется отсутствием семейной истории о нём. Синдром был обнаружен Джереми Оллгроувом и его коллегами в 1978 году. ААА представляет аббревиатуру синдрома ахалазии-аддисонианизма-алакримии. Алакримия является обычно ранним проявлением. Это прогрессирующее заболевание, которое может длиться годами, чтобы развить полноценную клиническую картину.
Синдром Коккейна , также называемый синдром Нил-Дингуолл — редкое аутосомно-рецессивное, нейродегенеративное расстройство, характеризующееся недостатком роста, нарушением развития нервной системы, аномальной чувствительностью к солнечному свету (фотосенсибилизация), заболеваниями глаз и преждевременным старением. Нездоровый вид и неврологические расстройства являются критериями для диагностики, а светочувствительность, нарушения слуха и ненормальные глаза — другие весьма общие черты. Возможны проблемы любого или всех внутренних органов. Это связано с группой расстройств, называемых лейкодистрофия. В основе расстройства лежит дефект механизма репарации ДНК. Интересно, в отличие от других дефектов репарации ДНК, пациенты с CS не предрасположены к раку или инфекции. Синдром Коккейна редок, но это разрушительная болезнь, которая, как правило, приводит к смерти в первом или втором десятилетии жизни. Мутация специфических генов в синдроме Коккейна известна, но широко распространенные эффекты и его отношения с репарацией ДНК еще не очень хорошо исследованы.
Синдром Пирсона — тяжелое врожденное аутосомно-рецессивное заболевание, вызванное мутацией гена, ответственного за кодирование β2-цепи ламинина. На долю которого приходится около 2,5% нефротического синдрома в течение первого года жизни, обычно клинически проявляющегося в течение первых 3 месяцев. В России зафиксирован единичный случай этого заболевания.
Дефицит WNT4 — редкое генетическое заболевание, которое затрагивает только женщин. Оно приводит к недоразвитию, а иногда и полному отсутствию матки и влагалища. Дефицит WNT4 вызван мутациями гена WNT4. При этом заболевании в крови пациенток обнаруживается аномально высокий уровень андрогенов, что может послужить причиной развития вторичных мужских половых признаков, таких как рост волос на лице и груди по мужскому типу. У женщин с этим заболеванием отсутствуют менструальные циклы, но молочные железы развиваются по женскому типу. Дефицит WNT4 связан, но отличается от Синдрома Майера — Рокитанского — Кустера — Хаузера. У некоторых женщин, у которых был первоначальный диагноз МРКХ, в дальнейшем диагностируют дефицит WNT4. Большинство женщин с синдромом МРКХ не имеют генетических мутаций гена WNT4.
Синдро́м Ло́уренса-Му́на. Редкое аутосомно-рецессивное генетическое заболевание, характеризующиеся ожирением, умственной отсталостью, атаксией, дистрофией сетчатки, гипопитуитаризмом, большим количеством пальцев рук или ног. Синдром связан с нарушением функции гипоталамических центров.

