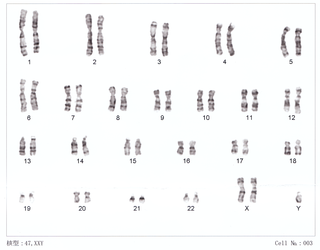
Синдром 48, XXYY
| Синдром 48, XXYY | |
|---|---|
 | |
| МКБ-10-КМ | Q98.8 |
| МКБ-9-КМ | 758.81[1] |
Синдром 48, XXYY — это аномалия хромосом, при которой у человека имеется дополнительная X и Y-хромосома. Клетки человека обычно содержат две половые хромосомы, одну от матери и одну от отца. Обычно женщины имеют две Х-хромосомы (XX), а мужчины имеют одну Х и одну Y-хромосому (XY). Появление по крайней мере одной Y-хромосомы с правильно функционирующим геном SRY делает человека физиологически мужчиной. Следовательно, люди с XXYY являются генотипически мужчинами. Мужчины с синдромом XXYY имеют 48 хромосом вместо типичных 46. По оценкам, он случается в 1 случае на каждые 18 000-40 000 новорожденных[2].
Симптомы
Симптомы этого синдрома включают в себя:
- Задержки развития
- Нарушение речи
- Трудности с обучаемостью
- Симптомы СДВГ
- Сколиоз
- Клинодактилия
- Низкий мышечный тонус
- Плоскостопие
- Крипторхизм
- Низкий уровень тестостерона
Причины
Обычно люди имеют 46 хромосом в каждой клетке. Две из 46 хромосом, известные как X и Y, называются половыми хромосомами, потому что они помогают определить, будут ли у человека развиваться мужские или женские половые признаки. Женщины обычно имеют две Х-хромосомы (46, XX), а мужчины имеют одну Х-хромосому и одну Y-хромосому (46, XY). Синдром 48, XXYY возникает из-за наличия дополнительной копии обеих половых хромосом в каждой из клеток. Дополнительные копии генов на Х-хромосоме препятствуют половому развитию мужчины, препятствуя нормальному функционированию яичек и снижая уровень тестостерона. Многие гены обнаруживаются только в X или Y хромосоме, но гены в областях, известных как псевдоаутосомные области, присутствуют в обеих половых хромосомах. Дополнительные копии генов из псевдоаутосомных областей лишних X и Y хромосом вносят вклад в симптомы синдрома; однако конкретные гены не были идентифицированы[3][4].
Генетика
Синдром не наследуется; Он обычно возникает случайно при формировании репродуктивных клеток (яйцеклетки и сперма). Ошибка в клеточном делении, называемая недисключением, приводит к появлению репродуктивной клетки с аномальным количеством хромосом. При синдроме 48, XXYY, дополнительные половые хромосомы почти всегда происходят из сперматозоидов. Несоответствие может привести к тому, что сперматозоид приобретет две дополнительные половые хромосомы, в результате чего сперматозоид будет иметь три половые хромосомы (одну Х и две Y-хромосомы). Если этот сперматозоид оплодотворяет нормальную яйцеклетку одной Х-хромосомой, у получающегося ребёнка будет две Х-хромосомы и две Y-хромосомы в каждой из клеток его тела.
В небольшом проценте случаев синдром 48, XXYY является следствием непереключения половых хромосом у эмбриона 46, XY очень скоро после оплодотворения. Это означает, что нормальный сперматозоид с одной Y-хромосомой оплодотворил нормальную яйцеклетку с одной X-хромосомой, но сразу после оплодотворения, несопряжение половых хромосом заставило эмбрион получить две дополнительные половые хромосомы, в результате чего зародыш имеет кариотип 48, XXYY[3].
Диагностика
Для диагностики проверяют кариотип[5].
Лечение
Пациенты обычно должны наблюдаться у эндокринолога. При наличии гипогонадизма лечение тестостероном следует рассматривать у всех людей, независимо от когнитивных способностей, из-за положительного влияния на здоровье костей, мышечный тонус, усталость и выносливость, а также с возможными преимуществами для психического здоровья / поведения[2].
У большинства детей с XXYY наблюдаются некоторые задержки в развитии и трудности в обучении. Следовательно, эти аспекты должны наблюдаться: психология (когнитивное и социально-эмоциональное развитие), логопедия, трудотерапия и физиотерапия. Следует организовать консультации с развивающими педиатрами, психиатрами или неврологами для разработки плана лечения, включающего терапию, поведенческие вмешательства, образовательную поддержку и психотропные препараты для поведенческих и психиатрических симптомов. Общие диагнозы, такие как неспособность к обучению, СДВГ, расстройства аутистического спектра, перепады настроения, тиковые расстройства и другие проблемы психического здоровья, должны быть рассмотрены, обследованы и пролечены. В этой группе наблюдаются хорошие реакции на стандартные медикаментозные методы лечения от невнимательности, импульсивности, тревоги и нестабильности настроения, и такое лечение может положительно влиять на успеваемость, эмоциональное благополучие в долгосрочной перспективе. Плохая координация мелкой моторики может сделать письмо медленным и трудоемким занятием, и трудотерапия и клавиатура должны быть введены в раннем возрасте, чтобы облегчить школьную работу и навыки самопомощи. Образовательные трудности должны оцениваться с помощью полной психологической оценки, чтобы выявить расхождения между устными и служебными навыками и выявить индивидуальные академические потребности. Языковые навыки часто страдают в течение всей жизни, и в зрелом возрасте могут потребоваться вмешательства в области логопедической терапии, направленные на развитие выразительных языковых навыков, диспраксии и др. Адаптивные навыки представляют собой затруднения, требующие поддержки на уровне сообщества почти для всех людей в зрелом возрасте[2]. Могут потребоваться дополнительные рекомендации по лечению, основанные на индивидуальных сильных и слабых сторонах синдрома XXYY[6].
Прогноз
Пациенты имеют практически нормальную продолжительность жизни, но им требуются регулярное медицинское наблюдение[3][7].
История
Первое опубликованное сообщение о мальчике с кариотипом 48, XXYY было опубликовано Сильфестом Малдалом и Чарльзом Оки в Манчестере, в 1960 году[8]. Он был описан у 15-летнего мальчика с психическими расстройствами, у которого были признаки синдрома Клайнфелтера; однако, тест на кариотип показал 48, XXYY. Из-за этого, синдром 48, XXYY изначально считался разновидностью синдрома Клайнфелтера. Общие физические и медицинские особенности, обусловленные наличием дополнительной Х-хромосомы, включают высокий рост, развитие дефицита тестостерона в подростковом и / или зрелом возрасте (гипергонадотропный гипогонадизм) и бесплодие. Тем не менее, недавние исследования выявили некоторые важные различия у людей с кариотипом 48, XXYY по сравнению с 47, XXY[5]. Наиболее важные различия обусловлены влиянием дополнительных X и Y-хромосом на развитие нервной системы, что приводит к более высокой частоте задержек развития в раннем детстве, неспособности к обучению или умственной отсталости, трудностей с адаптивным функционированием, расстройств нервного развития, таких как СДВГ или расстройства аутистического спектра и психологические/поведенческие проблемы, включая тревогу, депрессию и нарушение регуляции настроения. Кроме того, больший процент мужчин с XXYY имеют дополнительные медицинские проблемы, такие как судороги, врожденные аномалии локтевого сустава (радиульный синостоз) и тремор по сравнению с мужчинами с XXY. XXYY до сих пор считается вариацией синдрома Клайнфелтера по некоторым определениям, главным образом потому, что патофизиология дисфункции яичек не отличается от 47, XXY, и самые последние исследования не предполагают, что должны быть какие-либо различия в оценке и лечение дефицита тестостерона в 48, XXYY по сравнению с 47, XXY[8]. Однако для психологических и поведенческих симптомов синдрома XXYY обычно требуются более обширные оценки, вмешательства и поддержка по сравнению с 47, XXY из-за более сложного вовлечения в развитие нервной системы. Наблюдается значительная вариабельность между людьми по количеству и серьёзности медицинских проблем и проблем развития нервной системы, связанных с XXYY, и у некоторых людей наблюдаются легкие симптомы, в то время как другие страдают более значительным образом[2].
Примечания
- ↑ Monarch Disease Ontology release 2018-06-29 — 2018-06-29 — 2018.
- ↑ 1 2 3 4 Tartaglia N, Davis S, Hench A, et al. (June 2008). «A New Look at XXYY Syndrome: Medical and Psychological Features». Am. J. Med. Genet. A. 146A (12): 1509-22. doi:10.1002/ajmg.a.32366. PMC 3056496. PMID 18481271.
- ↑ 1 2 3 48,XXYY syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program. rarediseases.info.nih.gov. Дата обращения: 9 июня 2019. Архивировано 16 апреля 2021 года.
- ↑ Genetics Home Reference. 48,XXYY syndrome (англ.). Genetics Home Reference. Дата обращения: 9 июня 2019. Архивировано 12 июня 2019 года.
- ↑ 1 2 University of California - UC Newsroom | Researchers define characteristics, treatment options for XXYY Syndrome. web.archive.org (1 апреля 2010). Дата обращения: 9 июня 2019. Архивировано 1 апреля 2010 года.
- ↑ Visootsak J, Rosner B, Dykens E, Tartaglia N, Graham JM, Jr (2007). «Behavioral phenotype of sex chromosome aneuploidies: 48, XXYY, 48, XXXY, and 49,XXXXY». Am. J. Med. Genet. A. 143A (11): 1198—1203. doi:10.1002/ajmg.a.31746. PMID 17497714.
- ↑ INSERM US14-- ALL RIGHTS RESERVED. Orphanet: 48,XXYY syndrome (англ.). www.orpha.net. Дата обращения: 9 июня 2019. Архивировано 29 июля 2018 года.
- ↑ 1 2 Cotran, Ramzi S.; Kumar, Vinay; Fausto, Nelson; Nelso Fausto; Robbins, Stanley L.; Abbas, Abul K. (2005). Robbins and Cotran pathologic basis of disease. St. Louis, Mo: Elsevier Saunders. p. 179. ISBN 978-0-7216-0187-8.