Синдром поликистозных яичников — полиэндокринный синдром, сопровождающийся нарушениями функции яичников, поджелудочной железы, коры надпочечников, гипоталамуса и гипофиза.

Синдро́м нечувстви́тельности к андроге́нам — врожденные эндокринные вариации полового развития, вызванные мутацией гена, отвечающего за андрогеновый рецептор. Подобные синдромы варьируются в зависимости от структуры и чувствительности аномального рецептора. Клинические фенотипы находятся в диапазоне от номинального мужского телосложения с мягким синдромом сперматогенеза до крайне феминного телосложения, в дополнение к наличию Y-хромосомы.

Синдро́м Ма́ртина — Белл — наследственное заболевание.
Иммунодефици́ты — нарушения иммунологической реактивности, обусловленные выпадением одного или нескольких компонентов иммунного аппарата или тесно взаимодействующих с ним неспецифических факторов.

Синдро́м гиперкортици́зма — предклиническое состояние, при котором происходит длительное хроническое воздействие на организм избыточного количества гормонов коры надпочечников, независимо от причины, которая вызвала повышение количества этих гормонов в крови.
Синдром Торга-Винчестера — синдром, впервые описанный в 1969 году; у носителей которого отмечается спектр проявлений, в том числе сниженный рост вследствие патологии костей и суставов, помутнения роговицы, грубые черты лица, подкожные узелковые утолщения, огрубение кожи, гипертрихоз. Отклонения вызываются разнообразными мутациями гена MMP2. Их разнообразие привело к тому, что они были описаны как три различных синдрома: синдром Торга, синдром Винчестера, NAO-синдром, и лишь в 2006 году объединены под новым общим именем.

Синдром MERRF — редкое митохондриальное заболевание, вызываемое мутациями в следующих генах: MTTK, MTTL1, MTTH, MTTS1, MTTS2, MTTF. Симптомы MERRF также проявляются при мутациях гена MTND5.
Синдром Вискотта — Олдрича — редкое Х-сцепленное рецессивное заболевание, характеризующееся наличием экземы, тромбоцитопении, иммунодефицита, и кровавого поноса. Синоним — синдром экземы-тромбоцитопении-иммунодефицита в соответствии с оригинальным описанием Олдрича, сделанным в 1954 году.
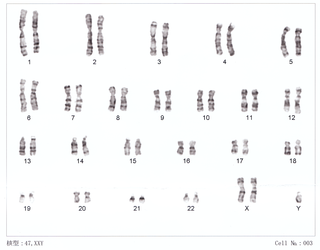
Синдром Клайнфельтера — генетическое отклонение. Клиническая картина синдрома описана в 1942 году в работах Гарри Клайнфельтера и Фуллера Олбрайта. Генетической особенностью этого синдрома является разнообразие цитогенетических вариантов и их сочетаний (мозаицизм). Обнаружено несколько типов полисомии по хромосомам X и Y у лиц мужского пола: 47, XXY; 47, XYY; 48, XXXY; 48, XYYY; 48 XXYY; 49 XXXXY; 49 XXXYY. Наиболее распространён синдром Клайнфельтера. Общая частота его колеблется в пределах 1 на 500—700 новорождённых мальчиков, что делает данный синдром первым по частоте встречаемости среди хромосомных болезней.
Синдром Свайера, XY дисгенезия гонад, женская гонадальная дисгенезия или гонадальная дисгенезия — генетическое нарушение, вариант гипогонадизма с кариотипом 46,XY. Организм человека с синдромом Свайера имеет характерный для мужского организма набор хромосом, но половые железы представляют собой гонадный тяж и не производят гормоны. В результате он имеет женские гениталии, женскую репродуктивную систему и выглядит как женщина. В период полового созревания развитие вторичных половых признаков не происходит и наблюдается аменорея. Существует практика удаления гонад в раннем возрасте с целью предотвращения развития рака.

Синдро́м Ба́рттера — форма гиперальдостеронизма с гиперплазией юкстагломерулярного аппарата почек и резистентностью к сосудосуживающему действию ангиотензина II, обусловленной внешними (вторичными) нарушениями передачи сигнала ангиотензина II.
Синдро́м де ля Шапе́ля — назван в честь исследователя, впервые охарактеризовавшего его в 1972 году. Синдром относится к редкой хромосомной патологии, возникающей в результате кроссинговера между X- и Y-хромосомами в процессе мейоза, в результате чего одна или обе X-хромосомы содержат нормальный мужской ген SRY. Распространённость данного синдрома составляет 4—5 на 100 000, что меньше встречаемости синдрома Клайнфельтера.
Синдром Макла — Уэльса (MWS) — это мутация в гене CIAS1 с развитием холодового аутовоспалительного синдрома. Является редким аутовоспалительным заболеванием наследственного характера. Преимущественно этническая распространённость — народы Северной Европы. Тип наследования — аутосомно-доминантный. Генетическая основа — мутация гена CIAS1, расположенного на длинном плече 1-й хромосомы (1q44) и кодирующего белка криопирина. Данный белок является основной образуемого в клетке супрамолекулярного комплекса, называемого инфламмасомой, выполняющего функцию превращения pro-IL-1β в активную форму, а также принимающего участие в выполнении программы апоптоза. Холодовой аутовоспалительный синдром тесно связан с двумя другими синдромами: семейной холодовой крапивницы и мультисистемным воспалительным заболеванием неонатального возраста — фактически, все связаны с мутациями гена. В целом Синдром Макла — Уэльса относится к группе криопирин-связанных периодических синдромов (CAPS).
PTEN — фосфатаза с двойной субстратной специфичностью, продукт гена PTEN. Субстратами этой фосфатазы могут быть как белки, так и фосфатидилинозитол-3-фосфаты. PTEN катализирует отщепление фосфатной группы в положении 3D инозитольного кольца фосфатидилинозитол-3-фосфатов, лишая их таким образом функций вторичных посредников при передаче сигнала в клетке. Эта фосфатаза является одним из немногих негативных регуляторов PI3K/AKT/mTOR-сигнального пути, что делает её антионкобелком. Ген PTEN часто бывает мутирован при различных типах злокачественных опухолей.

X-сцепленное рецессивное наследование — один из видов сцепленного с полом наследования. Такое наследование характерно для признаков, гены которых расположены на Х-хромосоме и которые проявляются только в гомозиготном или гемизиготном состоянии. Такой тип наследования имеет ряд врождённых наследственных заболеваний у человека, эти заболевания связаны с дефектом какого-либо из генов, расположенных на половой Х-хромосоме, и проявляются в случае, если нет другой Х-хромосомы с нормальной копией того же гена. В литературе встречается сокращение XR для обозначения X-сцепленного рецессивного наследования.
Синдром Ушера — сравнительно редкое генетическое заболевание, вызываемое мутацией одного из 10 генов, приводящее к врождённой нейросенсорной тугоухости и прогрессирующей потере зрения. Одна из основных причин слепоглухоты. В настоящее время неизлечим. Наследуется по аутосомно-рецессивному принципу.

Синдром Дуэйна — врождённый редкий тип косоглазия, чаще всего характеризуется неспособностью глаза двигаться наружу. Синдром был впервые описан офтальмологами Якобом Штиллингом (1887) и Зигмундом Тюрком (1896), а впоследствии стал носить имя Александра Дуэйна, который осветил этот синдром более подробно в 1905 году.
Синдро́м Ке́рнса — Се́йра (англ. Kearns–Sayre syndrome, сокращённо KSS) — митохондриальная миопатия с типичным началом до 20-летнего возраста. KSS является более серьёзным синдромным вариантом хронической прогрессирующей внешней офтальмоплегии, синдром, который характеризуется изолированным поражением мышц, контролирующих движения век и контролирующих движения глаз. Это приводит к птозу и офтальмоплегии соответственно. KSS включает в себя триаду уже описанных CPEO, двустороннюю пигментную ретинопатию и блокаду сердца. Другие области участия могут включать в себя мозжечковую атаксию, проксимальную мышечную слабость, глухоту, сахарный диабет, дефицит гормона роста, гипопаратиреоз или другие эндокринные нарушения. В обоих этих заболеваниях вовлечение мышц может начаться односторонним, но всегда развивается в двусторонний дефицит, который является прогрессирующим.
Дефицит WNT4 — редкое генетическое заболевание, которое затрагивает только женщин. Оно приводит к недоразвитию, а иногда и полному отсутствию матки и влагалища. Дефицит WNT4 вызван мутациями гена WNT4. При этом заболевании в крови пациенток обнаруживается аномально высокий уровень андрогенов, что может послужить причиной развития вторичных мужских половых признаков, таких как рост волос на лице и груди по мужскому типу. У женщин с этим заболеванием отсутствуют менструальные циклы, но молочные железы развиваются по женскому типу. Дефицит WNT4 связан, но отличается от Синдрома Майера — Рокитанского — Кустера — Хаузера. У некоторых женщин, у которых был первоначальный диагноз МРКХ, в дальнейшем диагностируют дефицит WNT4. Большинство женщин с синдромом МРКХ не имеют генетических мутаций гена WNT4.
Синдром Смит — Магенис (СМС) — наследственное заболевание, имеет такие проявления, как умственная отсталость, аномалии лица, проблемы со сном и многочисленные поведенческие проблемы, в том числе и такие, как самоповреждение. Частота синдрома Смит — Магенис составляет 1 случай на 15 000-25 000 человек. Синдром Смит — Магенис, как правило, обусловлен микроделецией в коротком плече 17-й хромосомы (17p), и иногда его называют 17p-синдром.