Синтез олигонуклеотидов


Синтез олигонуклеотидов — это химический синтез относительно коротких фрагментов нуклеиновых кислот с заданной химической структурой (последовательностью). Метод применяется в современной лабораторной практике для получения олигонуклеотидов нужной последовательности быстрым и недорогим способом.
Наиболее распространённый способ синтеза олигонуклеотидов основан на использовании амидофосфитов — строительных блоков, которые являются реакционноспособными производными дезоксирибонуклеозидов (dA, dC, dG, T) или рибонуклеозидов (A, C, G, U) и позволяют синтезировать короткие фрагменты ДНК и РНК соответственно. Применяются также и другие амидофосфиты, позволяющие вводить в цепь продукта модифицированные нуклеозиды, различные метки и функциональные группы. В ходе синтеза эти реагенты поочерёдно, в порядке, задаваемом последовательностью целевого продукта, присоединяют к растущей на твердофазном носителе цепи. Этот процесс был полностью автоматизирован в конце 1970-х годов и в настоящее время выполняется в специальных синтезаторах, управляемых компьютерами.
После завершения синтеза олигонуклеотид отделяют от носителя, удаляют защитные группы, использовавшиеся во время синтеза и очищают полученный продукт при помощи электрофореза или ВЭЖХ.
Синтетические олигонуклеотиды находят широкое применение в молекулярной биологии и медицине, например, как антисмысловые олигонуклеотиды, праймеры для секвенирования и амплификации ДНК, зонды для определения комплементарных последовательностей ДНК и РНК, инструменты для нацеленного введения мутаций и сайтов рестрикции, а также для синтеза искусственных генов.
История
В процессе эволюции синтеза олигонуклеотидов было разработано четыре основных метода создания связей между нуклеозидами. Эти методы детально рассмотрены в подробных обзорах литературы[1][2][3].
Ранние работы и современный H-фосфонатный метод
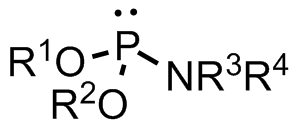
В начале 1950-х годов группа Александра Тодда из Кембриджа заложила основу для H-фосфонатного и фосфотриэфирного методов синтеза олигонуклеотидов[4][5], синтезировав защищённый хлорфосфат 4, введя его в реакцию с 3′-защищённым тимидином 5 и подтвердив структуру полученного защищённого динуклеотида 6 при помощи ферментативного расщепления. Как ни странно, дальнейшая работа в этом направлении в Кембридже не велась (Тодд отмечал в своей автобиографии, что синтез олигонуклеотидов его не привлекал)[1].
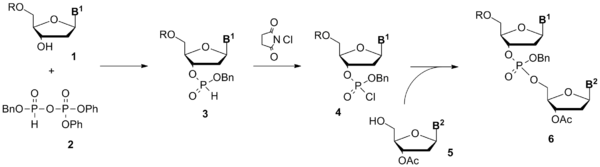



Возвращение к работам Тодда произошло только тридцать лет спустя, когда две исследовательские группы адаптировали H-фосфонатную конденсацию к твердофазному синтезу, используя нуклеозидные H-фосфонаты в качестве строительных блоков, а пивалоилхлорид, 2,4,6-триизопропилбензолсульфонилхлорид (TIPS-Cl) и другие соединения — в качестве активаторов[6][7]. Практически метод был организован в виде простого синтетического цикла, состоящего из двух стадий: снятия диметокситритильной защиты и конденсации.
Окисление H-фосфонатной диэфирной связи между нуклеозидами в данном методе проводят после синтеза олигонуклеотидной цепи под действием раствора йода в водном пиридине. В случае необходимости, окисление может проводиться в безводных средах[8]. Метод также позволяет синтезировать тиофосфатные[9] и селенофосфатные[10] аналоги олигонуклеотидов. Окисление тетрахлорметаном в присутствии первичных и вторичных аминов приводит к амидофосфатным аналогам[11][12].
Как правило, 5′-гидроксильная группа во всех 3′-Н-фосфонатах нуклеозидов и аминогруппа в Н-фосфонатах нуклеозидов c основаниями A, G и С защищаются перед использованием в синтезе теми же группами, что и в амидофосфитном методе. Однако, защита аминогрупп не является строго обязательной[8][13].
Фосфодиэфирный метод
В 1950-х годах Хар Гобинд Корана и сотрудники разработали фосфодиэфирный метод, в котором 3′-O-ацетилнуклеозид-5′-O-фосфат активировался N,N'-дициклогексилкарбодиимидом (DCC) или п-толуолсульфонилхлоридом (TsCl), а затем вводился в реакцию с 5′-O-защищённым нуклеозидом с образованием динуклеозидмонофосфата[14]. После удаления защитной ацетильной группы в основной среде, проводили дальнейшее удлинение цепи. Пошаговой конденсацией были синтезированы наборы три- и тетрамерных олигонуклеотидов. Кроме того осуществлялась конденсации олигомерных фрагментов с целью получения более длинных олигонуклеотидов[1].
Защита фосфатных групп в фосфодиэфирном синтезе не использовалась, а именно это, по-видимому, приводило к побочным реакциям и снижало выход синтеза. Однако, Корана считал, что постановка защиты на фосфатные группы приведёт к потере главного достоинства олигонуклеотидов — их полиэлектролитной природы, которую, как он полагал, можно эффективно использовать для очистки олигонуклеотидов. Важной находкой Кораны стало использование тритильных защитных групп для защиты 5′-гидроксильных нуклеозидов[1].
Фосфотриэфирный метод
В 1960-х годах группы под руководством Р. Летсингера[нем.] (англ. R. Letsinger)[15][16] и К. Риза (англ. C. Reese)[17] разработали фосфотриэфирный метод. Определяющее отличие от фосфодиэфирного подхода состоит в предварительной защите фосфата в реагенте и продукте цианэтильной группой -CH2CH2CN. Это изменение исключило возможность образования олигонуклеотидов с разветвлением у фосфатных групп. Бо́льшая селективность метода позволила использовать более реакционноспособные конденсирующие реагенты и катализаторы[18][19], которые значительно уменьшили продолжительность синтеза. Фосфотриэфирный метод был реализован как в растворе, так и в твердофазном варианте[1].
Данным методом удалось синтезировать два двухцепочечных олигонуклеотида длиной 77 и 104 пары оснований, последовательность которых соответствовала А- и В-цепям инсулина, что впоследствии позволило успешно экспрессировать эти гены[1].
Фосфитный триэфирный метод
В 1970-х годах в практику был введен фосфиттриэфирный метод образования межнуклеозидных связей, в котором были использованы существенно более реакционноспособные нуклеозидные производные на основе P(III), первоначально хлорфосфиты[20]. Позже группа под руководством М. Карузерса (англ. M. Caruthers) использовала менее агрессивные и более селективные 1H-тетразолидофосфиты и реализовала метод в твердофазном варианте[21]. Вскоре после этого сотрудники из той же группы улучшили метод путём использования в качестве строительных блоков более стабильных нуклеозидных амидофосфитов. Замена менее практичной метильной защитной группы фосфата[22][23][24] на более удобную 2-цианэтильную группу[25] дала тот вариант нуклеозидных амидофосфитов, который является стандартом реагентов для синтеза олигонуклеотидов и в настоящее время. Многочисленные дальнейшие усовершенствования методов синтеза мономерных блоков, олигонуклеотидных синтезаторов и протоколов сборки и деблокирования превратили амидофосфитный подход в очень надёжный и производительный метод получения синтетических олигонуклеотидов[26].
Синтез амидофосфитным методом
Строительные блоки
Нуклеозидные амидофосфиты
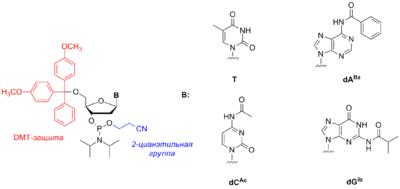
В 1976 году Летсингер и сотрудники обнаружили, что соединения трёхвалентного фосфора являются гораздо более активными реагентами, чем соответствующие производные пятивалентного фосфора. Роль соединений P(III) стала очевидной после разработки М. Карузерсом N,N-диизопропиламидофосфитных производных нуклеозидов (нуклеозидных амидофосфитов)[1][22], которые и сегодня выполняют роль стандартных строительных блоков в амидофосфитном методе синтеза. Для предотвращения нежелательных побочных реакций функциональные группы амидофосфитов должны быть заблокированы защитными группами. После окончания сборки олигонуклеотидной цепи все защитные группы удаляются, что приводит к целевому олигонуклеотиду. Ниже рассмотрены защитные группы применяемые в стандартных коммерчески доступных нуклеозидных амидофосфитах[27]:
- Гидроксильная группа в 5′-положении защищается 4,4'-диметокситритильной (DMT) защитной группой, удаляемой в кислой среде.
- Тимин и урацил, азотистые основания тимидина и уридина соответственно, не имеют экзоциклических аминогрупп, поэтому не нуждаются в защитных группах. Гуанин имеет экзоциклическую аминогруппу с низкой основностью, поэтому он не вступает в побочные реакции с амидофосфитами в условиях реакции конденсации. Однако, амидофосфитный реагент на основе N2-незащищенного 5′-O-DMT-2′-дезоксигуанозина плохо растворим в ацетонитриле — растворителе, наиболее часто используемом в синтезе олигонуклеотидов. Напротив, N2-защищённые производные того же соединения хорошо растворимы в ацетонитриле, поэтому и применяются гораздо шире. Азотистые основания аденин и цитозин содержат аминогруппы, способные реагировать с амидофосфитами в процессе синтеза, и поэтому для синтеза в стандартных условиях эти группы необходимо защищать. Тем не менее, путём введения в синтетический цикл дополнительных стадий можно добиться использования и незащищённых амидофосфитов dA и dC[28]. Защитные группы на азотистых основаниях должны сохраняться в течение всего синтеза, поэтому используются такие группы, реакционная способность которых противоположна реакционной способности 4,4'-диметокситритильной группы, которая удаляется после каждого цикла. Чаще всего используются два подхода, описанных ниже.
- В первом, стандартном, подходе для защиты аденина и цитозина используется бензоильная защита (Bz), тогда как гуанин защищаются изобутирильной группой (iBu)[29]. Позже для защиты цитозина была предложена ацетильная (Ac) группа, которая может быть удалена аммиаком, или смесью аммиака с метиламином[30].
- Во второй, более мягкой, защитной схеме аденин защищают изобутирильной[31] или феноксиацетильной (PAC)[32] группами. Цитозин содержит ацетильную защитную группу[30], а гуанин защищён 4-изопропилфеноксиацетильной (iPr-PAC)[33] или диметилформамидиновой (dmf)[34] группами. Мягкие защитные группы удаляются легче стандартных, однако, амидофосфиты с данными группами менее стабильны при хранении в растворе.

- Фосфитная группа защищается 2-цианэтильной группой[25]. Присутствие фосфитной защиты обязательно для амидофосфита и новообразованной фосфиттриэфирной группы до окисления последней в фосфотриэфир. В то же время, присутствие защиты на фосфатных группах уже собранного олигонуклеотидного фрагмента не является обязательным для успешного проведения дальнейших циклов конденсации[35].
- В синтезе РНК используются строительные блоки с «дополнительной» 2′-гидроксильной группой, не участвующей в синтезе. Её защищают трет-бутилдиметилсилильной (TBDMS)[36][37][38] или триизопропилсилилоксиметильной (TOM)[39][40] группой. Обе группы удаляются действием фторид-иона.
- Фосфитная группа также содержит диизопропиламинную группу (i-Pr2N), реакционноспособную в кислых условиях. При активации под действием кислотного катализатора эта группа замещается 5′-гидроксильной группой растущей цепи[22].
Ненуклеозидные амидофосфиты


Ненуклеозидные амидофосфиты — это амидофосфитные реагенты, разработанные для введения разнообразных функциональных групп и меток в концевое положение олигонуклеотида или между нуклеотидными остатками посреди последовательности. Чтобы быть пригодным для введения в середину цепи, амидофосфит должен содержать, по крайней мере, две гидроксильные группы, одна из которых защищается DMT-группой, а вторая несёт реакционноспособную амидофосфитную группировку.
Ненуклеозидные амидофосфиты применяются для введения в олигонуклеотид различных групп, которые не встречаются в природных нуклеозидах. Синтезирован широкий спектр подобных реагентов, служащих, например, для введения 5′-фосфата[41], аминогруппы[42], меркаптогруппы[43], альдегидной[44] и карбоксильной[45] групп, алкиновых фрагментов[46], флуоресцентных красителей[47] и тушителей, гидрофильных[48] и гидрофобных[49] модификаций, биотина[50] и др.
Синтетический цикл
Синтез олигонуклеотидов проводится путём пошаговой конденсации строительных блоков к 5′-концу растущей цепи, пока не будет собрана целевая последовательность. Протекание побочных реакций накладывает ограничение на длину синтезируемого олигонуклеотида (до 200 нуклеотидных остатков), поскольку число ошибок накапливается с увеличением длины целевого продукта[26]. Совокупность операций, необходимых для удлинения цепи на один нуклеотидный остаток, называется циклом синтеза и состоит из четырёх химических реакций.
Стадия 1: Удаление тритильной защиты
Защитная группа DMT удаляется раствором кислоты, например, 2%-ой трихлоруксусной кислотой или 3%-ой дихлоруксусной кислотой в инертном растворителе (хлористом метилене или толуоле). Образующийся диметокситритильный катион оранжевого цвета вымывается из системы. В результате образуется закреплённый на твердофазном носителе предшественник олигонуклеотида со свободной 5′-гидроксильной группой. Проведение процесса в течение более длительного времени или с использованием более концентрированных растворов кислоты приводит к побочной реакции депуринизации — отщепления пуриновых оснований от остатка рибозы.
Стадия 2: Конденсация
Раствор нуклеозидного амидофосфита (0,02—0,2 М) или смеси нескольких амидофосфитов в ацетонитриле активируют 0,2—0,7 М раствором кислотного катализатора на основе азола: 1H-тетразола, 2-этилтиотетразола[51], 2-бензилтиотетразола[52], 4,5-дицианимидазола[53] и др. Смешение компонентов происходит в коммуникационных линиях синтезатора в процессе доставки реагентов в реактор, содержащий твердофазный носитель. Активированный амидофосфит в 1,5—20-кратном избытке вводится во взаимодействие с 5′-гидроксильной группой, образуя фосфитную триэфирную связь. Конденсация амидофосфитов 2′-дезоксинуклеозидов протекает очень быстро и в небольшом масштабе занимает, как правило, около 20 секунд. Пространственно-затрудненные амидофосфиты рибонуклеозидов реагируют значительно медленнее (5—15 мин)[54][55][56][57]. Реакция весьма чувствительна к присутствию воды, особенно при использовании разбавленных растворов амидофосфитов, поэтому она проводится в безводном растворителе, обычно, ацетонитриле. При увеличении масштабов синтеза используют меньшие избытки и более концентрированные растворы амидофосфитов. После завершения реакции избыток реагентов и побочные продукты удаляются из реактора промыванием.
Стадия 3: Кэпирование
Кэпирование проводится путём обработки твердофазного носителя смесью уксусного ангидрида и 1-метилимидазола (реже — DMAP) в качестве катализатора. В рамках амидофосфитного синтеза эта стадия служит двум целям:
- После завершения стадии конденсации небольшая доля 5′-гидроксильных групп (0,1—1 %) остаётся непрореагировавшими и должна быть выведена из процесса дальнейшего удлинения цепи, чтобы предотвратить образование олигонуклеотидов с недостающими нуклеотидными остатками внутри цепи. С этой целью оставшиеся гидроксильные группы защищаются ацетильными группами, устойчивыми к действию растворов кислот, используемых для снятия DMT-защиты.
- Сообщалось также, что амидофосфиты, активированные 1H-тетразолом, с невысоким выходом реагируют с кислородом карбонильной группы в O6-положении гуанозина[58]. При окислении смесью йода с водой этот побочный продукт претерпевает отщепление пуринового основания. Образующиеся апуриновые сайты легко гидролизуются в ходе конечного снятия защитных групп олигонуклеотида, что приводит к образованию двух более коротких олигонуклеотидов и уменьшению выхода целевого продукта. O6-модификации быстро удаляются под действием кэпирующего реагента, если кэпирование проводится перед стадией окисления.
Стадия 4: Окисление
Полученная в результате конденсации межнуклеозидная трехкоординированная фосфитная группа не является природной и обладает ограниченной стабильностью в условиях синтеза. Обработка носителя йодом и водой в присутствии слабого основания (пиридин, 2,6-лутидин или коллидин) окисляет фосфит в фосфотриэфир, предшественник природной фосфодиэфирной межнуклеозидной связи. Окисление можно проводить и в безводных условиях с использованием трет-бутилгидропероксида[59] или более удобного реагента, (1S)-(+)-(10-камфорсульфонил)оксазиридина[60].
Твердофазные носители
В процессе всего твердофазного синтеза олигонуклеотид ковалентно связан с твердофазным носителем через 3′-гидроксильную группу. Носитель обычно помещается в колонки, размер которых зависит от масштаба синтеза. В последние 10 лет получили широкое распространение высокопроизводительные олигонуклеотидные синтезаторы, в которых носитель помещается в лунки планшета (96 или 384 лунок на планшет)[61]. После окончания синтеза олигонуклеотид отщепляется от носителя и переходит в раствор.
Материал носителя
| Размер пор CPG, Å[62] | Загрузка, мкмоль/г | Длина продукта, оснований |
|---|---|---|
| 500 | 80—90 | <50 |
| 1000 | 50—60 | 80 |
| 1500 | 35—45 | 100 |
| 2000 | 20—30 | 150 |
| 3000 | 200 |
В отличие от органического твердофазного синтеза и пептидного синтеза, синтез олигонуклеотидов лучше протекает на ненабухающих или слабонабухающих твердофазных носителях. Наиболее часто употребимыми носителями являются стекло с контролируемым размером пор (англ. controlled pore glass, CPG) и макропористый полистирол (MPPS)[63].
- Главной характеристикой CPG является диаметр пор, который может быть равным от 70 до 4000 Å, при этом поры весьма однородны по размеру (отклонение составляет ±10 % для 80 % пор). Для того, чтобы сделать такой носитель пригодным для синтеза, его обрабатывают (3-аминопропил)триэтоксисиланом[англ.] (APTES), получая аминопропильное CPG. Также часто используются CPG c удлинённым спейсером (англ. long chain aminoalkyl CPG, LCAA CPG)[63]. От размера пор зависят две ключевые характеристики синтеза: масштаб, определяемый числом активных функциональных групп на единицу массы носителя, и максимальная длина синтезируемого олигонуклеотида. Данные по наиболее распространёнными носителям CPG приведены в таблице[62].
- Также в синтезе олигонуклеотидов применяется макропористый полистирол, получаемый сополимеризацией стирола и дивинилбензола, с введённой аминометильной модификацией. Такой полистирол позволяет синтезировать олигонуклеотиды длиной менее 40—50 оснований. Для небольших синтезов можно использовать полистирольный носитель с загрузкой от 10 до 45 мкмоль/г. Для синтезов в масштабе 25—100 мкмоль используется полистирол с загрузкой 200 до 400 мкмоль/г. Наконец, полистирол больше, чем CPG, подходит для крупномасштабных синтезов (более 100 мкмоль)[62].
Химия линкера




Для нужд олигонуклеотидного синтеза к аминогруппам аминопропильного CPG, LCAA CPG или аминометильного MPPS ковалентно присоединяют нуклеозидные сукцинаты или ненуклеозидные линкеры. Обычно используют три различных типа носителей.
- Нуклеозидные носители. В исторически первом подходе синтез олигонуклеотида проводится на носителе, к которому заранее через фрагмент янтарной кислоты ковалентно присоединён 3′-концевой, стартовый, нуклеозид. Соответственно, синтез начинается с присоединения амидофосфита, соответствующего не первому, а второму нуклеотиду, считая с 3′-конца. Недостатком такого носителя является то, что перед началом синтеза необходимо выбрать вариант нуклеозидного носителя, соответствующий 3′-концевому нуклеозиду в синтезируемом олигонуклеотиде, что уменьшает производительность синтетического процесса и увеличивает вероятность человеческой ошибки[64]. Предложены также альтернативные твердофазные носители со спейсером на основе дигликолевой и щавелевой кислот для более быстрого отделения продукта от носителя[63].
- Универсальные носители. В данном методе синтез начинается с универсального носителя, к которому присоединён ненуклеозидный линкер[65]. Амидофосфит, соответствующий 3′-терминальному нуклеозиду, присоединяют к универсальному носителю по стандартной методике в ходе первого синтетического цикла. После этого продолжают сборку требуемой последовательности, а затем олигонуклеотид отщепляют с поверхности носителя. Преимущество данного подхода заключается в том, что во всех синтезах независимо от того, какую последовательность необходимо синтезировать, может быть использован единственный универсальный носитель[64].
- Специальные носители используются для присоединения некоторой функциональной или репортерной группы к 3′-положению синтетических олигонуклеотидов. Коммерчески доступны носители для введения аминогрупп[66], меркаптогрупп[67], тушителей флуоресценции[68] и др.
Тиофосфатные олигонуклеотиды и их синтез
Тиофосфатные олигонуклеотиды — это модифицированные олигонуклеотиды, в которых один из атомов кислорода в фосфатном остатке замещен на атом серы. Широко используются только те тиофосфаты, в которых сера не является связующим звеном между нуклеозидным остатком и атомом фосфора. В этом случае замена кислорода на серу приводит к образованию нового центра хиральности на атоме P(V), поэтому в простейшем случае динуклеотида образуются SP- и RP-диастереомеры. В n-мерном олигонуклеотиде, в котором все (n-1) межнуклеотидных связей являются тиофосфатными, число диастереомеров составляет 2(n-1). Будучи неприродными аналогами нуклеиновых кислот, олигонуклеотидные тиофосфаты значительно более устойчивы к гидролизу нуклеазами, классом ферментов, расщепляющих нуклеиновые кислоты путём гидролиза P-O-связи фосфодиэфирного мостика. Это свойство определяет использование тиофосфатов в качестве антисмысловых олигонуклеотидов в приложениях in vivo и in vitro, где неизбежно воздействие нуклеаз. Подобным образом, чтобы увеличить стабильность малых интерферирующих РНК, часто вводят, по крайней мере, одну тиофосфатную связь в 3′-положение смысловой и антисмысловой цепи. В оптически чистых олигонуклеотидных тиофосфатах диастереомеры, в которых все фосфорные центры имеют SP-конфигурацию, более устойчивы к ферментативному гидролизу, чем их RP-аналоги. Однако, синтез оптически чистых тиофосфатов сложен[69][70].
Синтез олигонуклеотидных тиофосфатов аналогичен синтезу природных олигонуклеотидов. Различие заключается в том, что стадия окисления заменяется реакцией сульфурирования. Для введения серы применяются следующие коммерчески доступные реагенты:
- 3-(Диметиламинометилиденамино)-3H-1,2,4-дитиазол-3-тион (DDTT) обеспечивает высокую скорость сульфурирования и обладает стабильностью в растворе[71].
- Реагент Бокажа (англ. Beaucage Reagent) имеет более высокую растворимость в ацетонитриле и обеспечивает протекание реакции за более короткое время. Однако, он имеет ограниченную стабильность в растворе и менее эффективен при сульфурировании связей РНК[72].
- N,N,N',N'-Тетраэтилтиурамдисульфид (TETD) растворим в ацетонитриле, однако, реакция сульфурирования межнуклеозидной связи ДНК занимает 15 мин, что в 10 раз медленнее, чем в случае двух предыдущих соединений[73].
Разработаны также другие сульфурирующие реагенты[74].
В синтезе тиофосфатных олигонуклеотидов стадия кэпирования проводится после сульфурирования. Если кэпирования проводить перед сульфурированием, то после кэпирования твердофазный носитель может содержать остаточные количества уксусного ангидрида и 1-метилимидазола. Кэпирующая смесь затрудняет реакцию переноса серы, что приводит к образованию тиофосфатных олигонуклеотидов с повышенным содержанием фосфатных звеньев. В таком случае рекомендуют проводить кэпирование после реакции сульфурирования[71].
Автоматизация


Ранее олигонуклеотидный синтез проводился вручную в растворе или на твёрдой фазе. Для твердофазного синтеза были приспособлены различные устройства на основе шприцев с пористыми мембранами или миниатюрных стеклянных фильтров, позволяющие промывать твердофазный носитель растворами реагентов и удалять их при помощи вакуума[76].
В настоящее время твердофазный синтез проводится автоматически в олигонуклеотидных синтезаторах, управляемых компьютерами. Данные приборы состоят из серии клапанов, соединённых с ёмкостями для реагентов, а также соленоидами, которые открывают или закрывают тот или иной клапан. В свою очередь, соленоиды управляются компьютером, выполняющим программу синтеза. Когда клапан открыт, через колонку, содержащую твердофазный носитель, в течение времени, задаваемого программой, прокачивается определённый реагент. Время и последовательность подачи реагентов постоянны для каждого синтетического цикла; переменным является лишь мономерный реагент, который должен конденсироваться в данном цикле. Его выбор также производится программой в соответствии с заданной олигонуклеотидной последовательностью[77].
Синтез может быть реализован в колоночном, планшетном или чиповом формате. Колоночный метод синтеза пригоден для исследований, или крупномасштабного синтеза этих полимеров, где не требуется высокая производительность их синтеза. Планшетный формат разработан специально для высокопроизводительного синтеза в малом масштабе для удовлетворения растущей потребности производства и науки в синтетических олигонуклеотидах. Коммерчески доступны различные виды синтезаторов[78][79][80].
Пост-синтетическая обработка
Для получения целевого олигонуклеотида необходимо удалить все защитные группы олигонуклеотида:
- 5′-концевую DMT-группу;
- ацильные защитные группы на азотистых основаниях;
- 2-цианэтильные группы, защищающие межнуклеозидные фосфатные группы.
5′-DMT-группа, как правило, удаляется раствором кислоты ещё в ходе автоматического синтеза. Отщепление олигонуклеотида от носителя, удаление защитных групп оснований и цианэтильных защитных групп обычно происходит одновременно под действием неорганических оснований или аминов. Обычно для этих целей применяют водный аммиак, раствор метиламина, их смеси[30], газообразный аммиак или метиламин[81], реже растворами других первичных аминов и щелочей при комнатной либо повышенной температуре. Такая обработка удаляет все защитные группы с 2′-дезоксиолигонуклеотидов, давая раствор конечного продукта. В случае 2′-O-силилзащищённых олигорибонуклеотидов добавляется стадия удаления силильных защитных групп под действием фторид-иона[82].
Применение данного метода ограничено тем, что при такой обработке в качестве побочного продукта образуется акрилонитрил. В условиях снятия защитных групп акрилонитрил может алкилировать азотистые основания, в основном, N3-положение тимина и урацила с образованием 2-цианэтильных производных по реакции Михаэля. Образования этих побочных продуктов можно избежать обработкой олигонуклеотидов, связанных с твердофазным носителем, растворами оснований в органическом растворителе, например, 50 % триэтиламином в ацетонитриле[83] или 10 % диэтиламином в ацетонитриле[84].
Незащищённый олигонуклеотид может использоваться без дальнейшей очистки или подвергаться дополнительной очистке одним из следующих методов[85].
- Наиболее грубым методом очистки олигонуклеотида является обессоливание — отделение низкомолекулярных примесей, образовавшихся при удалении защитных групп (бензамид, изобутирамид, ацетат аммония и др.). Для больших количеств олигонуклеотидов обессоливание удобно проводить осаждением в этанол: при этом продукт выпадает в осадок, а примеси и — в некоторых случаях — более короткие олигонуклеотиды остаются в растворе. Для малых количеств применяется гель-фильтрация[86].
- Очистка от укороченных олигонуклеотидов может быть эффективно произведена при помощи электрофореза в полиакриламидном геле, который основан на разделении олигонуклеотидов по размеру вследствие их различной подвижности в геле под действием электрического поля. После проведения электрофореза по поглощению ультрафиолетового излучения определяют фрагмент геля, в которой находится целевой олигонуклеотид, эту часть вырезают и выделяют очищенный олигонуклеотид вымачиванием или электроэлюцией[87].
- Наиболее полная очистка достигается при использовании высокоэффективной жидкостной хроматографии (ВЭЖХ). В данном методе разделение основано на гидрофобном (обращённофазовая ВЭЖХ) или зарядовом (ионообменная ВЭЖХ) взаимодействии олигонуклеотидов с сорбентом. Сила этого взаимодействия влияет на порядок и время элюции продукта из хроматографической колонки, что позволяет достаточно эффективно разделять продукты различной длины. Часто применяют альтернативный подход, при котором 5′-DMT-группа олигонуклеотида перед очисткой не удаляется. В этом случае за счёт присутствия гидрофобной защитной группы его гидрофобность и сила взаимодействия с гидрофобным сорбентом значительно увеличиваются, а хроматографическая подвижность снижается, что делает выделение продукта более эффективным. DMT-группа затем удаляется в кислой среде, а раствор упаривают и обессоливают[88].
Характеризация
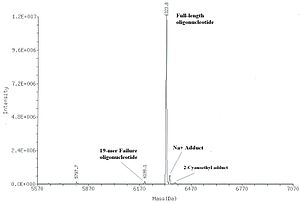
Как и в случае других органических веществ, целесообразно охарактеризовать олигонуклеотид после его получения. В более сложных случаях (исследование или крупномасштабный синтез) это делают дважды: после снятия защитных групп и после очистки. Несмотря на то, что наиболее корректным подходом к характеризации олигонуклеотида является секвенирование, относительно недорогая и рутинная процедура, экономические соображения препятствуют его введению в производство олигонуклеотидов. В ежедневной практике достаточно получить молекулярную массу олигонуклеотида путём регистрации его масс-спектра. Используется два метода масс-спектрометрии: электроспрей и МАЛДИ масс-спектрометрия. Для получения информативного спектра важно заменить все ионы металлов, которые могут присутствовать в образце, на ионы аммония или триалкиламмоиния.
- При ионизации электроспреем олигонуклеотид даёт набор ионов, которые соответствуют разной степени ионизации соединения. Олигонуклеотид с молекулярной массой М даёт ионы с массами (М — nH)/n, где М — молекулярная масса олигонуклеотида в кислотной форме (все отрицательные заряды фосфодиэфирных связей компенсированы наличием протонов), n — это степень ионизации, Н — атомная масса атома водорода (1 Да). Наиболее полезны для характеризации ионы с n от 2 до 5. Программное обеспечение, поставляемое с современными приборами, способно автоматически проводить поиск пиков ионов, принадлежащих определённому олигонуклеотиду, и вычислять молекулярную массу последнего.
- Для получения более детальной информации о примесях в олигонуклеотиде используются аналитические методы, комбинирующие ВЭЖХ и масс-спектрометрию[89] или капиллярный электрофорез и масс-спектрометрию[90].
См. также
Примечания
- ↑ 1 2 3 4 5 6 7 Reese C. B. Oligo- and poly-nucleotides: 50 years of chemical synthesis (англ.) // Org. Biomol. Chem. — 2005. — Vol. 3, no. 21. — P. 3851—3868. — doi:10.1039/b510458k. — PMID 16312051.
- ↑ Brown D. M. A Brief History of Oligonucleotide Synthesis // Protocols for Oligonucleotides and Analogs: Synthesis and Properties. — Springer, 1993. — P. 1—17. — (Methods in Molecular Biology). — doi:10.1385/0-89603-281-7:1.
- ↑ Iyer R. P., Beaucage S. L. 7.05 Oligonucleotide Synthesis // Comprehensive Natural Products Chemistry. — Elsevier, 1999. — P. 105—152.
- ↑ Michelson A. M., Todd A. R. Nucleotides part XXXII. Synthesis of a dithymidine dinucleotide containing a 3′: 5′-internucleotidic linkage (англ.) // J. Chem. Soc. — 1955. — P. 2632—2638. — doi:10.1039/JR9550002632.
- ↑ Hall R. H., Todd A., Webb R. F. 644. Nucleotides. Part XLI. Mixed anhydrides as intermediates in the synthesis of dinucleoside phosphates (англ.) // J. Chem. Soc. — 1957. — P. 3291—3296. — doi:10.1039/JR9570003291.
- ↑ Froehler B. C., Ng P. G., Matteucci M. D. Synthesis of DNA via deoxynucleoside H-phosphonate Intermediates (англ.) // Nucl. Acids Res. — 1986. — Vol. 14, no. 13. — P. 5399—5407. — doi:10.1093/nar/14.13.5399. Архивировано 6 мая 2022 года.
- ↑ Garegg P. J., Lindh I., Regberg T., Stawinski J., Strömberg R. Nucleoside H-phosphonates. III. Chemical synthesis of oligodeoxyribonucleotides by the hydrogenphosphonate approach (англ.) // Tetrahedron Lett. — 1986. — Vol. 27, no. 34. — P. 4051—4054. — doi:10.1016/S0040-4039(00)84908-4.
- ↑ 1 2 Wada T., Sato Y., Honda F., Kawahara S., Sekine M. Chemical Synthesis of Oligodeoxyribonucleotides Using N-Unprotected H-Phosphonate Monomers and Carbonium and Phosphonium Condensing Reagents: O-Selective Phosphonylation and Condensation // J. Amer. Chem. Soc. — 1997. — Т. 119, № 52. — С. 12710—12721. — doi:10.1021/JA9726015.
- ↑ Agrawal S., Tang J. Y. Efficient synthesis of oligoribonucleotide and its phosphorothioate analog using H-phosphonate approach // Tetrahedron Lett. — 1990. — Т. 31, № 52. — С. 7541—7544. — doi:10.1016/S0040-4039(00)97293-9.
- ↑ Tram K., Wang X., Yan H. Facile Synthesis of Oligonucleotide Phosphoroselenoates // Org. Lett. — 2007. — Т. 9, № 24. — С. 5103—5106. — doi:10.1021/ol702305v.
- ↑ Froehler B. C. Deoxynucleoside H-phosphonate diester intermediates in the synthesis of internucleotide phosphate analogs // Tetrahedron Lett. — 1986. — Т. 27, № 46. — С. 5575—5578. — doi:10.1016/S0040-4039(00)85269-7.
- ↑ Froehler B. C., Ng P. G., Matteucci M. D. Phosphoramidate analogs of DNA: synthesis and thermal stability of heteroduplexes // Nucl. Acids Res. — 1988. — Т. 16, № 11. — С. 4831—4839. — doi:10.1093/nar/16.11.4831.
- ↑ Kung P. P., Jones R. A. H-phosphonate DNA synthesis without amino protection // Tetrahedron Lett. — 1992. — Т. 33, № 40. — С. 5869—5872. — doi:10.1016/S0040-4039(00)61075-4.
- ↑ Gilham P. T., Khorana H. G. Studies on Polynucleotides. I. A New and General Method for the Chemical Synthesis of the C5″-C3″ Internucleotidic Linkage. Syntheses of Deoxyribo-dinucleotides (англ.) // J. Am. Chem. Soc. — 1958. — Vol. 80, no. 23. — P. 6212—6222. — doi:10.1021/ja01556a016.
- ↑ Letsinger R. L., Mahadevan V. Stepwise Synthesis of Oligodeoxyribonucleotides on an Insoluble Polymer Support (англ.) // J. Am. Chem. Soc. — 1966. — Vol. 88, no. 22. — P. 5319—5324. — doi:10.1021/ja00974a053. — PMID Ошибка выражения: неопознанное слово «ja» 10.1021/ja00974a053.
- ↑ Letsinger R. L., Ogilvie K. K. Nucleotide chemistry. XIII. Synthesis of oligothymidylates via phosphotriester intermediates (англ.) // J. Am. Chem. Soc. — 1969. — Vol. 91, no. 12. — P. 3350—3355. — doi:10.1021/ja01040a042.
- ↑ Reese C. B. The chemical synthesis of oligo- and poly-nucleotides by the phosphotriester approach (англ.) // Tetrahedron. — 1978. — Vol. 34, no. 21. — P. 3143—3179. — doi:10.1016/0040-4020(78)87013-6.
- ↑ Efimov V. A., Buryakova A. A., Reverdatto S. V., Chakhmakhcheva O. G., Ovchinnikov Yu. A. Rapid synthesis of long-chain deoxyribooligonucleotides by the N-methylimidazolide phosphotriester method (англ.) // Nucl. Acids Res. — 1983. — Vol. 11, no. 23. — P. 8369—8387. — doi:10.1093/nar/11.23.8369. — PMC 326588. Архивировано 6 мая 2022 года.
- ↑ Efimov V. A., Molchanova N. S., Chakhmakhcheva O. G. Approach to the Synthesis of Natural and Modified Oligonucleotides by the Phosphotriester Method Using O-Nucleophilic Intramolecular Catalysis (англ.) // Nucleosides, Nucleotides and Nucleic Acids. — 2007. — Vol. 26, no. 8—9. — P. 1087—1093. — doi:10.1080/15257770701516268. — PMID 18058542.
- ↑ Letsinger R. L., Finnan J. L., Heavner G. A., Lunsford W. B. Nucleotide chemistry. XX. Phosphite coupling procedure for generating internucleotide links (англ.) // J. Am. Chem. Soc. — 1975. — Vol. 97, no. 11. — P. 3278—3279. — doi:10.1021/ja00844a090. — PMID 1133350.
- ↑ Matteucci M. D., Caruthers M. H. Synthesis of deoxyoligonucleotides on a polymer support (англ.) // J. Am. Chem. Soc. — 1981. — Vol. 103, no. 11. — P. 3185—3191. — doi:10.1021/ja00401a041.
- ↑ 1 2 3 Beaucage S. L., Caruthers M. H. Deoxynucleoside phosphoramidites—A new class of key intermediates for deoxypolynucleotide synthesis (англ.) // Tetrahedron Lett. — 1981. — Vol. 22, no. 20. — P. 1859—1862. — doi:10.1016/S0040-4039(01)90461-7.
- ↑ McBride L. J., Caruthers M. H. Nucleotide chemistry. X. An investigation of several deoxynucleoside phosphoramidites useful for synthesizing deoxyoligonucleotides // Tetrahedron Lett. — 1983. — Т. 24, № 3. — С. 245—248. — doi:10.1016/S0040-4039(00)81376-3.
- ↑ Adams S. P., Kavka K. S., Wykes E. J., Holder S. B., Galluppi G. R. Hindered dialkylamino nucleoside phosphite reagents in the synthesis of two DNA 51-mers // J. Amer. Chem. Soc. — 1983. — Т. 105, № 3. — С. 661—663. — doi:10.1021/ja00341a078.
- ↑ 1 2 Sinha N. D., Biernat J., McManus J., Köster H. Polymer support oligonucleotide synthesis XVIII1.2): use of β-cyanoethyi-N,N-dialkylamino-/N-morpholino phosphoramidite of deoxynucleosides for the synthesis of DNA fragments simplifying deprotection and isolation of the final product (англ.) // Nucl. Acids Res. — 1984. — Vol. 12, no. 11. — P. 4539—4557. — doi:10.1093/nar/12.11.4539.
- ↑ 1 2 Beaucage S. L., Iyer R. P. Advances in the Synthesis of Oligonucleotides by the Phosphoramidite Approach (англ.) // Tetrahedron. — 1992. — Vol. 48, no. 12. — P. 2223—2311. — doi:10.1016/S0040-4020(01)88752-4.
- ↑ Glen Research. Applied Biosystems DNA & RNA Synthesizer Reagents (англ.). Дата обращения: 2 декабря 2012. Архивировано 16 января 2013 года.
- ↑ Gryaznov S. M., Letsinger R. L. Synthesis of oligonucleotides via monomers with unprotected bases (англ.) // J. Am. Chem. Soc. — 1991. — Vol. 113, no. 15. — P. 5876—5877. — doi:10.1021/ja00015a059.
- ↑ Virta P. Solid-phase synthesis of base-sensitive oligonucleotides (англ.) // ARKIVOC. — 2009. — P. 54—83. — ISSN 1551—7012. Архивировано 11 октября 2015 года.
- ↑ 1 2 3 Reddy M. P., Hanna N. B., Farooqui F. Ultrafast Cleavage and Deprotection of Oligonucleotides Synthesis and Use of CAc Derivatives (англ.) // Nucleosides and Nucleotides. — 1997. — Vol. 16, no. 7—9. — P. 1589—1598. — doi:10.1080/07328319708006236.
- ↑ McMinn D. L., Greenberg M. M. Synthesis of oligonucleotides containing 3′-alkyl amines using N-isobutyryl protected deoxyadenosine phosphoramidite (англ.) // Tetrahedron Lett. — 1997. — Vol. 38, no. 18. — P. 3123—3126. — doi:10.1016/S0040-4039(97)00568-6.
- ↑ Schulhof J. C., Molko D., Teoule R. The final deprotection step in oligonucleotide synthesis is reduced to a mild and rapid ammonia treatment by using labile base-protecting groups (англ.) // Nucl. Acids Res. — 1987. — Vol. 15, no. 2. — P. 397—416. — doi:10.1093/nar/15.2.397. — PMID 3822812. Архивировано 14 марта 2022 года.
- ↑ Zhu Q., Delaney M. O., Greenberg M. M. Observation and elimination of N-acetylation of oligonucleotides prepared using fast-deprotecting phosphoramidites and ultra-mild deprotection (англ.) // Bioorg. Med. Chem. Lett. — 2001. — Vol. 11, no. 9. — P. 1105—1107. — doi:10.1016/S0960-894X(01)00161-5. — PMID 11354354.
- ↑ McBride L. J., Kierzek R., Beaucage S. L., Caruthers M. H. Nucleotide chemistry. 16. Amidine protecting groups for oligonucleotide synthesis (англ.) // J. Am. Chem. Soc. — 1986. — Vol. 108, no. 8. — P. 2040—2048. — doi:10.1021/ja00268a052.
- ↑ Guzaev A. P., Manoharan M. Phosphoramidite Coupling to Oligonucleotides Bearing Unprotected Internucleosidic Phosphate Moieties (англ.) // J. Org. Chem. — 2001. — Vol. 66, no. 5. — P. 1798—1804. — doi:10.1021/jo001591e. — PMID 11262130.
- ↑ Ogilvie K. K., Theriault N., Sadana K. L. Synthesis of oligoribonucleotides (англ.) // J. Am. Chem. Soc. — 1977. — Vol. 99, no. 23. — P. 7741—7743. — doi:10.1021/ja00465a073.
- ↑ Usman N., Pon R. T., Ogilvie K. K. Preparation of ribonucleoside 3′-O-phosphoramidites and their application to the automated solid phase synthesis of oligonucleotides (англ.) // Tetrahedron Lett. — 1985. — Vol. 26, no. 38. — P. 4567—4570. — doi:10.1016/S0040-4039(00)98753-7.
- ↑ Scaringe S. A., Francklyn C., Usman N. Chemical synthesis of biologically active oligoribonucleotides using β-cyanoethyl protected ribonucleoside phosphoramidites (англ.) // Nucl. Acids Res. — 1990. — Vol. 18, no. 18. — P. 5433—5441. — doi:10.1093/nar/18.18.5433.
- ↑ Pitsch S., Weiss P. A., Wu X., Ackermann D., Honegger T. Fast and Reliable Automated Synthesis of RNA and Partially 2′-O- Protected Precursors (`Caged RNA') Based on Two Novel, Orthogonal 2′-O-Protecting Groups, Preliminary Communication (англ.) // Helv. Chem. Acta. — 1999. — Vol. 82, no. 10. — P. 1753—1761. — doi:10.1002/(SICI)1522-2675(19991006)82:10<1753::AID-HLCA1753>3.0.CO;2-Y.
- ↑ Pitsch S., Weiss P. A., Jenny L., Stutz A., Wu X. Reliable Chemical Synthesis of Oligoribonucleotides (RNA) with 2′-O-[(Triisopropylsilyl)oxy]methyl(2′-O-tom)-Protected Phosphoramidites (англ.) // Helv. Chem. Acta. — 2001. — Vol. 84, no. 12. — P. 3773—3795. — doi:10.1002/1522-2675(20011219)84:12<3773::AID-HLCA3773>3.0.CO;2-E.
- ↑ Guzaev A., Salo H., Azhayev A., Lönnberg H. A new approach for chemical phosphorylation of oligonucleotides at the 5′-terminus (англ.) // Tetrahedron. — 1995. — Vol. 51, no. 34. — P. 9375—9384. — doi:10.1016/0040-4020(95)00544-I.
- ↑ Sinha N. D., Cook R.M. The preparation and application of functionalised synthetic oligonucleotides: III. Use of H-phosphonate derivatives of protected amino-hexanol and mercapto-propanol or -hexanol (англ.) // Nucl. Acids Res. — 1988. — Vol. 16, no. 6. — P. 2659—2670. — doi:10.1093/nar/16.6.2659. Архивировано 6 мая 2022 года.
- ↑ Ede N. J., Tregear G. W., Haralambidis J. Routine Preparation of Thiol Oligonucleotides: Application to the Synthesis of Oligonucleotide-Peptide Hybrids (англ.) // Bioconjugate Chem. — 1994. — Vol. 5, no. 4. — P. 373—378. — doi:10.1021/bc00028a016. — PMID 7948105.
- ↑ Podyminogin M. A., Lukhtanov E. A., Reed M. W. Attachment of benzaldehyde-modified oligodeoxynucleotide probes to semicarbazide-coated glass (англ.) // Nucl. Acids Res. — 2001. — Vol. 29, no. 24. — P. 5090—5098. — doi:10.1093/nar/29.24.5090.
- ↑ Lebedev A. V., Combs D., Hogrefe R. I. Preactivated Carboxyl Linker for the Rapid Conjugation of Alkylamines to Oligonucleotides on Solid Support (англ.) // Bioconjugate Chem. — 2007. — Vol. 18, no. 5. — P. 1530—1536. — doi:10.1021/bc0603891. — PMID 17877414.
- ↑ Alvira M., Eritja R. Synthesis of Oligonucleotides Carrying 5′-5′ Linkages Using Copper-Catalyzed Cycloaddition Reactions (англ.) // Chem. Biodiversity. — 2007. — Vol. 4, no. 12. — P. 2798—2809. — doi:10.1002/cbdv.200790229. — PMID 18081090.
- ↑ Kvach M. V., Tsybulsky D. A., Ustinov A. V., Stepanova I. A., Bondarev S. L., Gontarev S. V., Korshun V. A., Shmanai V. V. 5(6)-Carboxyfluorescein Revisited: New Protecting Group, Separation of Isomers, and their Spectral Properties on Oligonucleotides (англ.) // Bioconjugate Chem. — 2007. — Vol. 18, no. 5. — P. 1691—1696. — doi:10.1021/bc7001874. — PMID 17696491.
- ↑ Jäschke A., Fürste J. P., Nordhoff E., Hillenkamp F., Cech D., Erdmann V. A. Synthesis and properties of oligodeoxyribonucleotide—polyethylene glycol conjugates (англ.) // Nucl. Acids Res. — 1994. — Vol. 22, no. 22. — P. 4810—4817. — doi:10.1093/nar/22.22.4810. — PMID 7984434.
- ↑ Musumeci D., Montesarchio D. Synthesis of a Cholesteryl-HEG Phosphoramidite Derivative and Its Application to Lipid-conjugates of the Anti-HIV 5′TGGGAG3′ Hotoda’s Sequence (англ.) // Molecules. — 2012. — Vol. 17. — P. 12378—12392. — doi:10.3390/molecules171012378. — PMID 23090019. Архивировано 2 ноября 2015 года.
- ↑ Kayushin A., Demekhina A., Korosteleva M., Miroshnikov A., Azhayev A. Synthesis of biotin-containing phosphoramidite linker with polyether spacer arm (англ.) // Nucleosides, Nucleotides and Nucleic Acids. — 2011. — Vol. 30, no. 7—8. — P. 490—502. — doi:10.1080/15257770.2011.587702. — PMID 21888541.
- ↑ Sproat B., Colonna F., Mullah B., Tsou D., Andrus A., Hampel A., Vinayak R. An Efficient Method for the Isolation and Purification of Oligoribonucleotides (англ.) // Nucleosides and Nucleotides. — 1995. — Vol. 14, no. 1—2. — P. 255—273. — doi:10.1080/15257779508014668.
- ↑ Welz R., Müller S. 5-(Benzylmercapto)-1H-tetrazole as activator for 2′-O-TBDMS phosphoramidite building blocks in RNA synthesis (англ.) // Tetrahedron Lett. — 2002. — Vol. 43, no. 5. — P. 795—797. — doi:10.1016/S0040-4039(01)02274-2.
- ↑ Vargeese C., Carter J., Yegge J., Krivjansky S., Settle A., Kropp E., Peterson K., Pieken W. Efficient activation of nucleoside phosphoramidites with 4,5-dicyanoimidazole during oligonucleotide synthesis (англ.) // Nucl. Acids Res. — 1998. — Vol. 26, no. 4. — P. 1046—1050. — doi:10.1093/nar/26.4.1046. Архивировано 6 мая 2022 года.
- ↑ Usman N., Ogilvie K. K., Jiang M. Y., Cedergren R. J. The automated chemical synthesis of long oligoribuncleotides using 2′-O-silylated ribonucleoside 3′-O-phosphoramidites on a controlled-pore glass support: synthesis of a 43-nucleotide sequence similar to the 3′-half molecule of an Escherichia coli formylmethionine tRNA // J. Amer. Chem. Soc. — 1987. — Т. 109, № 25. — С. 7845—7854. — doi:10.1021/ja00259a037.
- ↑ Ogilvie K. K., Usman N., Nicoghosian K., Cedergren R. J. Total chemical synthesis of a 77-nucleotide-long RNA sequence having methionine-acceptance activity // Proc. Natl. Acad. Sci. USA. — 1988. — Т. 85, № 16. — С. 5764—5768. — doi:10.1073/pnas.85.16.5764. — PMID 3413059. — PMC 281845.
- ↑ Wu T., Ogilvie K. K., Perreault J. P., Cedergren R. J. Convenient procedure for the preparation of specific mixed DNA-RNA polymers // J. Amer. Chem. Soc. — 1989. — Т. 111, № 22. — С. 8531—8533. — doi:10.1021/ja00204a043.
- ↑ Pon R. T. Enhanced coupling efficiency using 4-dimethylaminopyridine (DMAP) and either tetrazole, 5-(o-nitrophenyl)tetrazole, or 5-(p-nitrophenyl)tetrazole in the solid phase synthesis of oligoribonucleotides by the phosphoramidite procedure // Tetrahedron Lett. — 1987. — Т. 28, № 32. — С. 3643—3646. — doi:10.1016/S0040-4039(00)96344-5.
- ↑ Pon R. T., Usman N., Damha M. J., Ogilvie K. K. Prevention of guanine modification and chain cleavage during the solid phase synthesis of oligonucleotides using phosphoramidite derivatives (англ.) // Nucl. Acids Res. — 1986. — Vol. 14, no. 16. — P. 6453—6470. — doi:10.1093/nar/14.16.6453. — PMID 3748816. Архивировано 6 мая 2022 года.
- ↑ Alul R. H., Singman C. N., Zhang G., Letsinger R. L. Oxalyl-CPG: a labile support for synthesis of sensitive oligonucleotide derivatives // Nucl. Acids Res. — 1991. — Т. 19, № 7. — С. 1527—1532. — doi:10.1093/nar/19.7.1527. — PMID 2027761. Архивировано 6 мая 2022 года.
- ↑ New Product: 0.5M CSO for non-aqueous oxidation in DNA synthesis (англ.). Glenres.com. Дата обращения: 28 января 2013. Архивировано 4 февраля 2013 года.
- ↑ BioAutomation. DNA / RNA Oligonucleotide Synthesizer: MerMade 384. Дата обращения: 3 декабря 2012. Архивировано из оригинала 30 сентября 2011 года.
- ↑ 1 2 3 Guzaev A. P., Pon R. T. Attachment of Nucleosides and Other Linkers to Solid-Phase Supports for Oligonucleotide Synthesis // Current Protocols in Nucleic Acid Chemistry. — Wiley, 2013.
- ↑ 1 2 3 Pon R. T. Solid‐Phase Supports for Oligonucleotide Synthesis // Current Protocols in Nucleic Acid Chemistry. — Wiley, 2001.
- ↑ 1 2 Vaijayanthi B., Kumar P., Ghosh P. K., Gupta K. C. Recent advances in oligonucleotide synthesis and their applications (англ.) // Indian Journal of Biochemistry and Biophysics. — 2003. — Vol. 40, no. 6. — P. 377—391. — PMID 22900365. Архивировано 14 августа 2017 года.
- ↑ Guzaev A. P., Manoharan M. A Conformationally Preorganized Universal Solid Support for Efficient Oligonucleotide Synthesis (англ.) // J. Am. Chem. Soc. — 2003. — Vol. 125, no. 9. — P. 2380—2381. — doi:10.1021/ja0284613. — PMID 12603111.
- ↑ Petrie C. R., Reed M. W., Adams A. D., Meyer Jr. R. B. An improved CPG support for the synthesis of 3′-amine-tailed oligonucleotides (англ.) // Bioconjugate Chem. — 1992. — Vol. 3, no. 1. — P. 85—87. — doi:10.1021/bc00013a014. — PMID 1616954.
- ↑ Link Technologies. 3′-Thiol Modifier C3 S-S CPG. Дата обращения: 4 декабря 2012. Архивировано из оригинала 29 июня 2013 года.
- ↑ Lumiprobe. Black Hole 1 (BHQ-1) quencher CPG. Дата обращения: 4 декабря 2012. Архивировано из оригинала 23 ноября 2010 года.
- ↑ Wilk A., Grajkowski A., Phillips L. R., Beaucage S. L. Deoxyribonucleoside Cyclic N-Acylphosphoramidites as a New Class of Monomers for the Stereocontrolled Synthesis of Oligothymidylyl- and Oligodeoxycytidylyl- Phosphorothioates // J. Amer. Chem. Soc. — 2000. — Т. 122, № 10. — С. 2149—2156. — doi:10.1021/ja991773u.
- ↑ Lebedev A. V., Wickstrom E. The chirality problem in P-substituted oligonucleotides (англ.) // Perspectives in Drug Discovery and Design. — 1996. — Vol. 4, no. 1. — P. 17—40. — doi:10.1007/BF02172106.
- ↑ 1 2 Guzaev A. P. Reactivity of 3H-1,2,4-dithiazole-3-thiones and 3H-1,2-dithiole-3-thiones as sulfurizing agents for oligonucleotide synthesis (англ.) // Tetrahedron Lett. — 2011. — Vol. 52, no. 3. — P. 434—437. — doi:10.1016/j.tetlet.2010.11.086.
- ↑ Iyer R. P., Egan W., Regan J. B., Beaucage S. L. 3H-1,2-Benzodithiole-3-one 1,1-dioxide as an improved sulfurizing reagent in the solid-phase synthesis of oligodeoxyribonucleoside phosphorothioates (англ.) // J. Am. Chem. Soc. — 1990. — Vol. 112, no. 3. — P. 1253—1254. — doi:10.1021/ja00159a059.
- ↑ Vu H., Hirschbein B. L. Internucleotide phosphite sulfurization with tetraethylthiuram disulfide. Phosphorothioate oligonucleotide synthesis via phosphoramidite chemistry (англ.) // Tetrahedron Lett. — 1991. — Vol. 32, no. 26. — P. 3005—3008. — doi:10.1016/0040-4039(91)80672-S.
- ↑ Efimov V. A., Kalinkina A. L., Chakhmakhcheva O. G., Hill T. S., Jayaraman K. New efficient sulfurizing reagents for the preparation of oligodeoxyribonucleotide phosphorothioate analogues (ут) // Nucl. Acids Res. — 1995. — Т. 23, № 20. — С. 4029—4033. — doi:10.1093/nar/23.20.4029. — PMID 7479060.
- ↑ Ito H., Ike Y., Ikuta S., Itakura K. Solid phase synthesis of polynucleotides. VI. Farther studies on polystyrene copolymers for the solid support (англ.) // Nucl. Acids Res. — 1982. — Vol. 10, no. 5. — P. 1755—1769. — doi:10.1093/nar/10.5.1755.
- ↑ Tanaka T., Letsinger R. L. Syringe method for stepwise chemical synthesis of oligonucleotides (англ.) // Nucl. Acids Res. — 1982. — Vol. 10, no. 10. — P. 3249—3259. — doi:10.1093/nar/10.10.3249. — PMID 7099961. Архивировано 6 мая 2022 года.
- ↑ Alvarado-Urbina G., Sathe G. M., Liu W. C., Gillen M. F., Duck P. D., Bender R., Ogilvie K. K. Automated synthesis of gene fragments (англ.) // Science. — 1981. — Vol. 214, no. 4518. — P. 270—274. — doi:10.1126/science.6169150.
- ↑ BioAutomation. DNA / RNA Oligonucleotide Synthesizer: MerMade. Дата обращения: 11 декабря 2012. Архивировано 16 января 2013 года.
- ↑ БИОССЕТ. Дата обращения: 11 декабря 2012. Архивировано из оригинала 3 ноября 2012 года.
- ↑ Azco Biotech, Inc. Azco DNA/RNA Synthesizers. Дата обращения: 11 декабря 2012. Архивировано из оригинала 15 сентября 2011 года.
- ↑ Boal J. H., Wilk A., Harindranath N., Max E. E., Kempe T., Beaucage S. L. Cleavage of oligodeoxyribonucleotides from controlled-pore glass supports and their rapid deprotection by gaseous amines (англ.) // Nucl. Acids Res. — 1996. — Vol. 24, no. 15. — P. 3115—3117. — doi:10.1093/nar/24.15.3115. Архивировано 20 января 2016 года.
- ↑ Westman E., Strömberg R. Removal of t-butyldimethylsilyl protection in RNA-synthesis. Triethylamine trihydrofluoride (TEA, 3HF) is a more reliable alternative to tetrabutylammonium fluoride (TBAF) (англ.) // Nucl. Acids Res. — 1994. — Vol. 22, no. 12. — P. 2430—2431. — doi:10.1093/nar/22.12.2430. — PMID 7518583.
- ↑ Capaldi D. C., Gaus H., Krotz A. H., Arnold J., Carty R. L., Moore M. N., Scozzari A. N., Lowery K., Cole D. L., Ravikumar V. T. Synthesis of High-Quality Antisense Drugs. Addition of Acrylonitrile to Phosphorothioate Oligonucleotides: Adduct Characterization and Avoidance (англ.) // Org. Proc. Res. Dev. — 2003. — Vol. 7, no. 6. — P. 832—838. — doi:10.1021/op020090n.
- ↑ Glen Research. Deprotection — Volume 5 — On-Column Deprotection of Oligonucleotides in Organic Solvents. Дата обращения: 11 декабря 2012. Архивировано 16 января 2013 года.
- ↑ Applied Biosystems. Evaluating and Isolating Synthetic Oligonucleotides. The Complete Guide (2002). Дата обращения: 15 апреля 2013. Архивировано 19 апреля 2013 года.
- ↑ Evaluating and Isolating Synthetic Oligonucleotides, 2002, pp. 2—5—2—6.
- ↑ Evaluating and Isolating Synthetic Oligonucleotides, 2002, pp. 3—1—3—19.
- ↑ Evaluating and Isolating Synthetic Oligonucleotides, 2002, pp. 4—1—4—15.
- ↑ Krotz A. H., Gaus H., Hardee G. E. Formation of oligonucleotide adducts in pharmaceutical formulations // Pharmaceutical development and technology. — 2005. — Т. 10, № 2. — С. 283—90. — doi:10.1081/PDT-54464. — PMID 15926677.
- ↑ Willems A., Deforce D. L., Van Bocxlaer J. Analysis of oligonucleotides using capillary zone electrophoresis and electrospray mass spectrometry // Methods in Molecular Biology. — Springer, 2008. — Т. 384. Capillary Electrophoresis. — С. 401—414. — ISBN 978-1-59745-376-9. — doi:10.1007/978-1-59745-376-9_14.
Литература
Основные оригинальные работы
- Caruthers M. H. Chemical synthesis of DNA and DNA analogs (англ.) // Acc. Chem. Res. — 1991. — Vol. 24, no. 9. — P. 278—284. — doi:10.1021/ar00009a005.
- Letsinger R. L., Mahadevan V. Stepwise Synthesis of Oligodeoxyribonucleotides on an Insoluble Polymer Support (англ.) // J. Am. Chem. Soc. — 1966. — Vol. 88, no. 22. — P. 5319—5324. — doi:10.1021/ja00974a053. — PMID Ошибка выражения: неопознанное слово «ja» 10.1021/ja00974a053.
Обзоры
- Burtscher H., Berner S., Seibl R., Mühlegger K. Nucleic Acids // Ullmann's Encyclopedia of Industrial Chemistry. — Wiley, 2000. — doi:10.1002/14356007.a18_001.
- Iyer R. P., Beaucage S. L. 7.05 Oligonucleotide Synthesis // Comprehensive Natural Products Chemistry. — Elsevier, 1999. — P. 105—152.
- Reese C. B. Oligo- and poly-nucleotides: 50 years of chemical synthesis (англ.) // Org. Biomol. Chem. — 2005. — Vol. 3, no. 21. — P. 3851—3868. — doi:10.1039/b510458k. — PMID 16312051.
Методы
- Applied Biosystems. Evaluating and Isolating Synthetic Oligonucleotides. The Complete Guide (2002). Дата обращения: 15 апреля 2013. Архивировано 19 апреля 2013 года.
- Ch. 3. Synthesis of Unmodified Oligonucleotides // Current Protocols in Nucleic Acid Chemistry. — Wiley. — ISBN 9780471142706.
- Ch. 4. Synthesis of Modified Oligonucleotides and Conjugates // Current Protocols in Nucleic Acid Chemistry. — Wiley. — ISBN 9780471142706.
- Protocols for Oligonucleotides and Analogs: Synthesis and Properties / Editor: S. Agrawal. — Springer, 1993. — Т. 20. — (Methods in Molecular Biology). — ISBN 978-1-59259-507-5.