Система комплемента
Систе́ма комплеме́нта — комплекс защитных белков, постоянно присутствующих в крови. Это каскадная система протеолитических ферментов, предназначенная для гуморальной защиты организма от действия чужеродных агентов, она участвует в реализации иммунного ответа организма. Является важным компонентом как врождённого, так и приобретённого иммунитета. Выделяют три основных пути активации системы комплемента: классический, альтернативный и лектиновый. Для запуска классического пути комплемента необходима опсонизация чужеродной клетки антителами, а альтернативный и лектиновый пути могут активироваться в отсутствие антител. Поздние стадии у всех трёх путей активации системы комплемента одинаковы и включают образование мембраноатакующего комплекса, который нарушает целостность мембраны клетки-патогена и приводит к её гибели.
Система комплемента является эволюционно древним защитным механизмом, и некоторые её компоненты имеются даже у низших животных, таких как стрекающие. Многие патогены развили способность уклоняться от действия системы комплемента и стали устойчивы к ней. Недостаточность по многим компонентам комплемента или, наоборот, чрезмерная активность системы комплемента лежат в основе многих заболеваний человека.
Впервые система комплемента была описана в конце XIX века, а сам термин «комплемент» ввёл в употребление Пауль Эрлих.
Общая характеристика
Система комплемента состоит из поверхностных белков и белков плазмы крови, которые взаимодействуют друг с другом и с другими молекулами иммунной системы строго регулируемым образом, давая продукты, убивающие клетки патогенов. Белки комплемента являются белками плазмы крови, которые в состоянии покоя неактивны и активируются лишь при определённых условиях. Систему комплемента активируют микроорганизмы и антитела, прикреплённые к клеткам патогенов и другим антигенам. В ходе активации комплемента происходит несколько актов протеолиза, в результате которых образуются ферментативные комплексы с протеолитической активностью. Белки, приобретающие протеолитическую активность только после разрезания другими протеазами, называют зимогенами. Протеолитические каскады дают возможность постепенно усиливать изначальный сигнал, так как молекулы фермента, активированные на одном этапе, могут активировать больше молекул фермента на последующем этапе. Продукты активации системы комплемента ковалентно связаны с поверхностью микробных клеток, антителами, связанными с микробами, другими антителами, а также апоптотическими тельцами. Находясь в жидкой среде, белки комлемента остаются неактивными или активируются лишь на короткое время. После прикрепления к антигенам они становятся постоянно активными. Таким образом, система комплемента активируется и становится полностью функциональной только на поверхности клеток патогенов или местах, в которых присутствуют связанные с антигенами антитела. На нормальных клетках (но не клетках микробов) присутствуют регуляторные белки, подавляющие активацию системы комплемента, что обеспечивает защиту нормальных клеток организмов от её действия. Апоптотические тельца не имеют мембраносвязанных белков-ингибиторов комплемента и поэтому могут разрушаться системой комплемента, однако они способны поглощать белки-ингибиторы из крови[1].
В таблице ниже перечислены основные функции ключевых компонентов комплемента[2][3].
| Функция | Белки |
|---|---|
| Взаимодействие с комплексом антиген-антитело на поверхности микробной клетки | C1q[англ.] |
| Ферментативная активность по отношению к белкам системы комплемента | C1r[англ.], C1s[англ.], C2b, Bb, фактор D[англ.] |
| Опсонины и белки, связывающиеся с мембраной чужеродной клетки | C4b, C3b[англ.] |
| Медиаторы воспаления | C5a[англ.], C3a[англ.], C4a[англ.] |
| Белки, формирующие поры | C5[англ.], C6[англ.], C7[англ.], C8, C9[англ.] |
| Рецепторы комплемента[англ.] | CR1[англ.], CR2, CR3, CR4, CRIg[англ.] |
| Регуляторные белки комплемента | Ингибитор C1[англ.], C4BP[англ.], CR1, MCP, DAF, факторы H[англ.], I[англ.], CD59 |
Активация
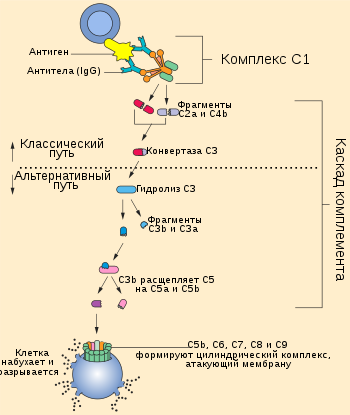
Существует три основных пути активации системы комплемента: классический путь, при котором комплемент активируют антитела некоторых изотипов[англ.], связанные с антигенами; альтернативный путь, при котором белки комплемента активируются на поверхности микробных клеток в отсутствие антител; лектиновый путь, при котором комплемент активируют лектины плазмы крови, связанные с остатками маннозы в составе полисахаридов на поверхности микроорганизмов. Классический путь был описан самым первым, однако альтернативный путь является более древним филогенетически и раньше появился в ходе эволюции. Хотя пути активации комплемента отличаются начальными этапами, все они приводят к образованию ферментативных комплексов, способных к расщеплению самого многочисленного белка комплемента — C3[англ.]. Альтернативный и лектиновый пути являются эффекторными механизмами врождённого иммунитета, а классический путь относят к числу гуморальных механизмов приобретённого (адаптивного) иммунитета[1].
Ключевое событие активации комплемента заключается в протеолизе белка комплемента C3 с образованием биологически активных продуктов, один из которых, C3b, далее ковалентно присоединяется к поверхности микробной клетки или антителу, связанному с антигеном. Важнейшую роль в активации комплемента играют два белковых комплекса: C3-конвертаза[англ.], которая расщепляет C3 на C3a[англ.] и C3b, и C5-конвертаза[англ.], разрезающая компонент комплемента C5 на C5a[англ.] и C5b. Белки, на которые расщепляются компоненты комплемента, принято обозначать строчными латинскими буквами, причём буквой a обозначается меньший фрагмент, а буквой b — больший. Все биологические функции комплемента зависят от протеолитического расщепления C3. В частности, C3b, ковалентно связанный с микробными клетками, стимулирует их фагоцитоз фагоцитами (нейтрофилами и макрофагами), которые экспрессируют рецепторы к C3b. Пептиды, образующиеся при разрушении C3 и других белков комплемента, стимулируют воспалительный ответ. Различия между путями активации комплемента заключаются в том, как образуется C3b, однако после разрушения C5 во всех трёх путях идут одни и те же реакции[4].
В таблице ниже перечислены основные стадии трёх путей активации комплемента[5].
| Стадия | Классический путь | Альтернативный путь | Лектиновый путь |
|---|---|---|---|
| Инициация | Узнавание комплекса антиген-антитело, связывание C1[англ.] | Спонтанная активация C3, образование и связывание Bb | Узнавание углеводов, образование комплекса MSP-MASP |
| Амплификация сигнала, образование C3-конвертазы | Расщепление и связывание C2 и C4[англ.], образование конвертазы C4bC2b | Активация C3 и фактора B[англ.], образование конвертазы C3bBb | Расщепление и связывание C2 и C4, образование конвертазы C4bC2b |
| Образование C5-конвертазы | Расщепление C3, образование C5-конвертазы | Стабилизация комплекса, образование C5-конвертазы | Расщепление C3, образование C5-конвертазы |
| Атака клеточной мембраны | Расщепление C5, образование мембраноатакующего комплекса | Расщепление C5, образование мембраноатакующего комплекса | Расщепление C5, образование мембраноатакующего комплекса |
Классический путь

Классический путь инициируется при связывании белка комплемента C1 с доменом CH2 молекулы иммуноглобулина G (IgG) или доменом CH3 молекулы иммуноглобулина M (IgM), которые уже связались с антигеном. Среди антител IgG классический путь наиболее эффективно активируют IgG3 и IgG1 (у человека). Белок C1 состоит из субъединиц C1q, C1r и C1s, причём C1q связывается с антителом, а C1r и C1s являются протеазами. C1q представляет собой гексамер, который специфически связывается с участками Fc тяжёлых цепей типа μ и некоторыми тяжёлыми цепями типа γ. Классический путь комплемента не может быть активирован свободными антителами, но только антителами, связанными с соответствующим антигеном, причём для активации необходимо, чтобы C1 связался с двумя и более участками Fc. Поскольку каждая молекула IgG имеет только один участок Fc, для связывания с C1 необходимо, чтобы две или более молекулы IgG находились рядом. Хотя свободные IgM в плазме крови являются пентамерами[англ.], путь комплемента не может быть активирован при связывании лишь с одной молекулой IgM, поскольку участки Fc каждого мономера располагаются так, что они не могут быть связаны одной молекулой C1. Одна молекула IgM, будучи пентамером, может связать две молекулы C1, поэтому IgM активирует комплемент эффективнее, чем IgG. C1r и C1s являются сериновыми протеазами и формируют тетрамер, в котором C1r и C1s имеются в числе двух молекул. Когда C1q связывается с IgG или IgM, связанный C1r активируется и вносит разрез в C1s, активируя его. Активированный C1s разрезает следующий белок каскада, C4, с образованием C4b. Как и C3b, C4b содержит внутреннюю тиоэфирную связь, которая обеспечивает ковалентное сшивание C4b с комплексом антиген-антитело на поверхности микробной клетки или непосредственно с поверхностью клетки. Следующий участник каскада, C2, связывается с C4b, ковалентно прикреплённым к поверхности клетки, и разрушается C1s с образованием фрагмента C2b, функции которого неизвестны. При этом C2a остаётся связанным с C4b на поверхности клетки патогена (в отличие от остальных компонентов комплемента, у C2 больший фрагмент называется C2a, а меньший фрагмент C2b, высвобождаемый при расщеплении С2, остаётся несвязанным). Комплекс C4b2a является C3-конвертазой и может связывать C3 и ферментативно расщеплять его. Связывание C3 с C3-конвертазой обеспечивается C4b, а C2a катализирует протеолиз C3. При расщеплении C3 образуются два фрагмента, меньший из которых, C3a, удаляется, а C3b может ковалентно связываться с белками клеточной поверхности или антителами, связанными с клеткой, на поверхности которой был активирован каскад комплемента. C3b также может взаимодействовать с фактором B и образовывать больше C3-конвертаз по альтернативному пути активации комплемента. Одна C3-конвертаза в итоге может давать начало сотням и тысячам молекул C3b на поверхности клетки, где был активирован комплемент. Ранние этапы классического и альтернативного путей комплемента имеют много похожего: C3 в альтернативном пути гомологичен C4 классического пути, а фактор B гомологичен C2. Некоторые молекулы C3b связываются с C3-конвертазой с образованием комплекса C4b2a3b, который является C5-конвертазой. C5-конвертаза расщепляет C5 и запускает поздние стадии каскада комплемента[6].
При инфекциях, вызванных пневмококками, запускается независимый от антител, но зависимый от C1 вариант классического пути, который активируется при связывании углеводов с лектинами на поверхности клетки. Некоторые макрофаги экспрессируют лектин C-типа, известный как SIGN-R1, который распознаёт полисахариды пневмококков и связывается с C1q. Благодаря этому активируется классический путь комплемента, в результате работы которого клетка пневмококка покрывается C3b[7].
Альтернативный путь
В отличие от классического пути, альтернативный путь системы комплемента не требует участия антител. В норме C3 постоянно расщепляется в плазме крови с низкой интенсивностью, и образующийся C3b может ковалентно связываться с белками на поверхности микробных клеток посредством домена, содержащего тиоэфирную связь, подобно C4b. Если C3b не связался с клеткой, то он подвергается быстрому гидролизу при участии той же тиоэфирной связи и инактивируется. C3b также имеет сайт связывания с белком плазмы крови фактором B. Фактор B связывается C3b, ковалентно сшитым с поверхностью микробной клетки, и расщепляется сериновой протеазой фактором D. Образовавшийся фрагмент Ba высвобождается, а более крупный фрагмент Bb остаётся связанным с C3b. Комплекс C3bBb является альтернативной C3-конвертазой и расщепляет дополнительные молекулы C3, обеспечивая амплификацию сигнала. C3b, полученный в ходе классического или лектинового путей, также может связываться с Bb с образованием комплекса, который расщепляет больше молекул C3. Если комплекс C3bBb формируется на поверхности клетки млекопитающего, то он быстро разрушается под действием регуляторных белков на поверхности клетки. Кроме того, на микробной клетке с комплексом C3bBb связывается белок комплемента пропердин, который стабилизирует комплекс; на клетках млекопитающих такого не происходит. Пропердин является единственным известным положительным регулятором комплемента. C3b и Bb могут формировать комплекс из двух молекул C3b и одной молекулы Bb, который функционирует как C5-конвертаза, которая расщепляет C5 и запускает поздние стадии каскада комплемента[8].
Лектиновый путь
Лектиновый путь активации комплемента не требует участия антител и запускается при связывании микробных полисахаридов лектинами, циркулирующими в плазме крови, такими как маннан-связывающий лектин (англ. mannan-binding lectin, MBL), или фиколинами[англ.]. MBL, L-фиколин и H-фиколин циркулируют в кровотоке, а M-фиколин выделяют активированные макрофаги в тканях. MBL связывается с остатками маннозы в составе полисахаридов, а фиколины связывают N-ацетилглюкозамин-содержащие гликаны. MBL и фиколины взаимодействуют с сериновыми протеазами группы MASP (от англ. MBL-associated serine proteases), которые структурно гомологичны C1r и C1s и выполняют аналогичные функции, а именно, расщепление C2 и C4 в ходе активации комплемента. Последующие стадии лектинового пути идентичны таковым в составе классического пути[7].
Поздние стадии

C5-конвертазы, образовавшиеся в ходе классического, альтернативного или лектинового пути, запускают последующие этапы каскада комплемента, кульминацией которых является образование мембраноатакующего комплекса. C5-конвертаза расщепляет C5 на высвобождаемый меньший фрагмент C5a и больший фрагмент C5b, который остаётся связанным с белками комплемента на поверхности микробной клетки. Последующие участники каскада комплемента — C6, C7, C8 и C9 — являются структурно близкими белками, лишёнными ферментативной активности. C5b временно сохраняет конформацию, в которой он может связать C6 и C7 с образованием комплекса C5b,6,7. C7 обладает гидрофобными свойствами и вставляется в липидный бислой клеточной мембраны, где становится высокоаффинным рецептором C8. Белок C8 имеет тримерную[англ.] структуру, и одна из его субъединиц связывается с комплексом C5b,6,7, при этом формируя ковалентную связь со второй субъединицей; третья субъединица интегрирует в клеточную мембрану. Получившийся в результате комплекс C5b,6,7,8 (C5b-8) обладает невысокой способностью к лизису клетки, и формирование полностью функционального мембраноатакующего комплекса завершается при связывании с C5b,6,7,8 компонента C9. C9 полимеризуется в местах взаимодействия с комплексом C5b,6,7,8 и формирует поры в мембране. Поры имеют диаметр около 100 ангстрем и формируют каналы, через которые свободно перемещаются вода и ионы. Вход воды в клетку из-за осмоса приводит к её набуханию и разрушению. Поры, сформированные C9, похожи на поры, образованные белком перфорином, который входит в состав гранул цитотоксических T-лимфоцитов и натуральных киллеров, кроме того, C9 структурно гомологичен перфорину[9].
Рецепторы комплемента
Многие функции комплемента опосредованы связыванием фрагментов комплемента с мембранными рецепторами, которые экспрессируются клетками разных типов. Основные сведения о главных группах рецепторов комплемента перечислены в таблице ниже[10].
| Группа рецепторов | Структура | Лиганды | Типы клеток | Функции |
|---|---|---|---|---|
| Рецепторы комплемента I типа (CR1, CD35) | Масса 160—250 кДа, содержат несколько повторов контрольных белков комплемента | C3b > C4b > iC3b[англ.] | Мононуклеарные фагоциты, нейтрофилы, T- и B-клетки, эритроциты, фолликулярные дендритные клетки | Фагоцитоз, уход иммунных комплексов[англ.] из кровотока, способствуют диссоциации C3-конвертазы, действуя как кофакторы разрушения C3b и C4a |
| Рецепторы комплемента II типа (CR2, CD21) | Масса 145 кДа, содержат несколько повторов контрольных белков комплемента | C3d, C3dg > iC3b | B-клетки, фолликулярные дендритные клетки, назофарингеальный эпителий | Корецепторы активации B-клеток, захватывание антигенов в герминальные центры, рецепторы для вируса Эпштейна — Барр |
| Рецепторы комплемента III типа (CR3, Mac-1, CD11bCD18) | Интегрины с α-субъединицей массой 165 кДа и β-субъединицей массой 95 кДа | iC3b, ICAM-1, связывают микробные клетки | Мононуклеарные фагоциты, нейтрофилы, натуральные киллеры | Фагоцитоз, адгезия лейкоцитов к эндотелию посредством ICAM-1 |
| Рецепторы комплемента IV типа (CR4, p150,95, CD11cCD18) | Интегрины с α-субъединицей массой 150 кДа и β-субъединицей массой 95 кДа | iC3b | Мононуклеарные фагоциты, нейтрофилы, натуральные киллеры | Фагоцитоз, адгезия клеток |
Рецепторы комлемента I типа (также известные как CR1 или CD35) преимущественно стимулируют фагоцитоз частиц, покрытых C3b и C4b, а также уход иммунных комплексов из кровотока. CR1 обладают высоким сродством к C3b и C4b и экспрессируются в основном клетками, происходящими из костного мозга: эритроцитами, нейтрофилами, моноцитами, макрофагами, эозинофилами, T- и B-клетками. Их также экспрессируют фолликулярные дендритные клетки, находящиеся в фолликулах периферических лимфоидных органов. CR1 на поверхности эритроцитов связывают циркулирующие по кровотоку иммунные комплексы, содержащие ковалентно связанные C3b и C4b, и перемещают их в печень и селезёнку. В этих органах фагоциты удаляют иммунные комплексы с поверхности эритроцитов, и эритроциты возвращаются в кровоток. CR1 также функционируют как регуляторы активации комплемента[10].
Рецепторы комплемента II типа (также известные как CR2 или CD21) стимулируют гуморальный иммунный ответ, усиливая активацию B-клеток антигенами и способствуя захвату комплексов антиген-антитело в герминальных центрах. CR2 присутствуют на поверхности B-лимфоцитов, фолликулярных дендритных клеток и некоторых эпителиальных клеток. Они связывают продукты расщепления C3b: C3d, C3dg и iC3b (i обозначает «неактивный» от англ. inactive). На B-клетках CR2 входят в состав тримолекулярных комплексов, включающих также нековалентно связанные белки CD19 и CD81. Этот комплекс обеспечивает усиление сигнала при связывании B-клетки с антигеном. CR2 на поверхности фолликулярных клеток захватывают комплексы антиген-антитело, покрытые iC3b и C3dg, в герминальные центры. У человека CR2 служат рецепторами вируса Эпштейна — Барр, который вызывает инфекционный мононуклеоз и некоторые онкологические заболевания. Вирус Эпштейн — Барр входит в B-клетки благодаря CR2[10].
Рецепторы комплемента III типа (CR3, Mac-1, CD11bCD18) представляют собой интегрины, которые служат рецепторами для iC3b, образующегося при расщеплении C3b. CR3 присутствуют на поверхности нейтрофилов, мононуклеарных фагоцитов, тучных клеток и натуральных киллеров. Рецептор CR3 состоит из двух нековалентно связанных субъединиц — α (CD11b) и β (CD18). На поверхности нейтрофилов и моноцитов рецепторы CR3 способствуют фагоцитозу микробных клеток, опсонизированных iC3b, кроме того, они могут непосредственно связываться с какими-то белками на поверхности бактериальных клеток, подлежащих фагоцитозу. Кроме того, рецепторы CR3 могут взаимодействовать с молекулами ICAM-1 на поверхности эндотелиальных клеток, облегчая адгезию лейкоцитов к эндотелию даже в отсутствие активации комплемента[11].
Рецепторы комплемента IV типа (CR4, p150,95, CD11cCD18) тоже являются интегринами, β-цепь которых идентична таковой у CR3, а α-субъединица называется CD11c. Рецепторы CR4 также распознают iC3b, и их функции сходны с таковыми у CR3. Они обильно экспрессируются дендритными клетками и являются молекулярным маркером[англ.] этого типа клеток[12].
На поверхности макрофагов в печени, известных как клетки Купфера, экспрессируются рецепторы комплемента из иммуноглобулинового семейства (CRIg). CRIg является интегральным мембранным белком, внеклеточная часть которого состоит из иммуноглобулиновых доменов. Рецепторы CRIg связывают C3b и iC3b и принимают участие в уничтожении опсонизированных бактерий[12].
Регуляция
Активация комплемента на поверхности клеток организма, а также слишком длительная активация комплемента на поверхности микробных клеток и комплексов антиген-антитело могут иметь губительные последствия для организма, поэтому активация каскада комплемента и стабильность активных белков комплемента жёстко регулируются посредством нескольких белков плазмы крови и белков, расположенных на поверхности клеток. Многие из этих белков-регуляторов, наряду с некоторыми компонентами классического и альтернативного путей, относятся к одному белковому семейству — семейству регуляторов активации комплемента, кодируются гомологичными генами, которые располагаются в геноме по соседству. Основные регуляторы активации комплемента перечислены в таблице ниже[12].
| Белок | Структура | Локализация | Взаимодействует с | Функции |
|---|---|---|---|---|
| Ингибитор C1 | Масса 140 кДа | Плазменный белок, концентрация 200 мкг/мл | C1r, C1s | Ингибитор сериновых протеаз, связывает C1r и C1s и вызывает их диссоциацию от C1q |
| Фактор I | Димер массой 88 кДа | Плазменный белок, концентрация 35 мкг/мл | C4b, C3b | Сериновая протеаза, расщепляет C3b и C4b, используя фактор H, MCP, C4BP и CR1 как кофакторы |
| Фактор H | Масса 150 кДа, содержит несколько повторов контрольных белков комплемента | Плазменный белок, концентрация 480 мкг/мл | C3b | Связывает C3b и вытесняет Bb, кофактор в реакции расщепления C3b, проводимого фактором I |
| C4-связывающий белок (C4BP) | Масса 570 кДа, содержит несколько повторов контрольных белков комплемента | Плазменный белок, концентрация 300 мкг/мл | C4b | Связывает C4b и вытесняет C2, кофактор в реакции расщепления C4b, проводимого фактором I |
| Мембранный кофактор (MCP, CD46) | Масса 45—70 кДа, содержит четыре повтора контрольных белков комплемента | Лейкоциты, эпителиальные клетки, эндотелиальные клетки | C3b, C4b | Кофактор в реакциях расщепления C4b и C3b, проводимых фактором I |
| Фактор ускорения разрушения (DAF) | Масса 70 кДа, GPI-якорь, содержит четыре повтора контрольных белков комплемента | Клетки крови, эпителиальные и эндотелиальные клетки | C4b2a, C3bBb | Вытесняет C2a из комплекса с C4b и Bb из комплекса с C3b (диссоциация C3-конвертаз) |
| CD59 | Масса 18 кДа, GPI-якорь | Клетки крови, эпителиальные и эндотелиальные клетки | C7, C8 | Блокирует связывание C9 и предотвращает образование мембраноатакующего комплекса |
Протеолитическая активность C1r и C1s подавляется плазменным белком, известным как ингибитор C1. Ингибитор C1 относится к ингибиторам сериновых протеаз группы серпинов, который мимикрирует[англ.] под нормальные субстраты C1r и C1s. После разрушения под действием C1r и C1s ингибитор C1 остаётся связанным с этими белками и вызывает их диссоциацию от C1q, тем самым останавливая классический путь активации комплемента. Таким образом, ингибитор C1 ограничивает количество активных комплексов C1r и C1s в плазме крови и ограничивает срок существования активных комплексов такого состава[13].
Несколько белков, локализующихся на поверхности клеток организма, связываются с C3b и C4b и не дают собраться C3- и C5-конвертазам на поверхности клеток. К числу таких отрицательных регуляторов, связывающихся с C3b клетки млекопитающего, относятся мембранный кофактор (MCP, или CD46), рецепторы комплемента I типа, фактор ускорения распада комплемента (англ. decay-accelerating factor, DAF), а также белок плазмы крови, известный как фактор H. C4b, прикрепившийся к поверхности клетки млекопитающего, связывают DAF, CR1, MCP, а также плазменный C4-связывающий белок (англ. C4 binding protein, C4BP). Связываясь с C3b или C4b, эти белки конкурентно подавляют[англ.] их связывание с компонентами C3-конвертазы. На поверхности микробных клеток таких белков нет, кроме того, по сравнению с клетками млекопитающих на клетках бактерий имеется меньше сиаловой кислоты, которая благоприятствует связыванию с поверхностью клетки ингибирующего регуляторного белка фактора B[14].
На поверхностях клеток организма-хозяина также имеется сериновая протеаза, известная как фактор I, которая расщепляет связавшийся с поверхностью C3b, но только в присутствии регуляторных белков-кофакторов, которыми служат MCP, фактор H, C4BP и CR1. В результате расщепления C3b, проводимого фактором I, образуются фрагменты iC3b, C3d и C3dg, которые не участвуют в активации комплемента, но распознаются рецепторами на фагоцитах и B-клетках[15].
Клетки организма многих типов экспрессируют поверхностный GPI-заякоренный белок CD59, препятствующий образованию мембраноатакующего комплекса. CD59 встраивается в состав собирающегося мембраноатакующего комплекса после сборки C5b-8, подавляя дальнейшее включение в его состав компонента C9. На поверхностях микробных клеток CD59 нет. Формированию мембраноатакующего комплекса также препятствует плазменный S-белок, который связывается с растворимыми комплексами C5b,6,7 и не даёт им встроиться в клеточную мембрану. Растущий мембраноатакующий комплекс может переместиться на мембрану другой клетки, отличной от той, на которой был активирован комплемент. Ингибиторы мембраноатакующего комплекса, расположенные на поверхности клеток организма-хозяина или циркулирующие в плазме крови, препятствуют его перемещения на другие клетки, не активировавшие комплемент[16].
Ингибиторы комплемента различаются по силе, которая зависит от многочисленности молекул ингибитора на поверхности клеток. Самым сильным ингибитором считают CD59, затем следуют DAF и MCP. При некоторых иммунологических заболеваниях работа регуляторных белков пересиливается чрезмерной активацией комплемента[17].
Функции
Функции системы комплемента в составе врождённого и адаптивного иммунного ответа состоят в стимуляции фагоцитоза микробных клеток, на поверхности которых активировался комплемент, воспаления и запуске лизиса клеток патогена. Фрагменты белков комплемента, образующиеся при его активации, облегчают активацию B-клеток и образование антител. Фагоцитоз, воспаление и стимуляцию гуморального иммунитета запускают протеолитические фрагменты белков комплемента, связывающиеся с рецепторами на клетках разных типов, а лизис инициирует мембраноатакующий комплекс[17].
Микробные клетки, на которых был активирован классический или альтернативный путь комплемента, покрываются C3b, iC3b и C4b, которые действуют как опсонины, и подвергаются фагоцитозу после связывания этих фрагментов со специфическими рецепторами на поверхности макрофагов и нейтрофилов. C3b и C4b связываются с CR1, а iC3b связывается с CR3 и CR4. В одиночку CR1 не может запускать фагоцитоз клеток, покрытых C3b, однако его способности к запуску фагоцитоза усиливаются, когда микробная клетка покрыта IgG. Стимулирующую роль по отношению к CR1-опосредованному фагоцитозу имеет интерферон γ, активирующий макрофаги. C3b- и iC3b-опосредованный фагоцитоз является важнейшим защитным механизмом врождённого и адаптивного иммунитета, особенно в случае бактерий, имеющих обогащённую полисахаридами капсулу, таких как пневмококки и менингококки[17].
Протеолитические фрагменты белков комплемента C5a, C4a и C3a запускают острое воспаление, активируя тучные клетки, нейтрофилы и клетки эндотелия. Связывание перечисленных пептидов с тучными клетками приводит к их дегрануляции и высвобождению вазоактивных[англ.] соединений, в числе которых гистамин. У нейтрофилов C5a стимулирует их подвижность, плотную адгезию с эндотелиальными клетками, а в высоких концентрациях стимулирует окислительный взрыв[англ.], в результате которого образуются активные формы кислорода. C5a также действует на эпителиальные клетки, повышая проницаемость эндотелия и экспрессию P-селектина на их поверхностях, что способствует связыванию с нейтрофилами. Действие C5a на тучные клетки, нейтрофилы и эндотелий способствует развитию воспаления в месте активации комплемента. C5a является самым сильным фактором дегрануляции тучных клеток, однако рецептор C5a, относящийся к группе GPCR, экспрессируют клетки разных типов: нейтрофилы, эозинофилы, базофилы, макрофаги, моноциты, тучные клетки, клетки эндотелия, гладких мышц, эпителиальные клетки и астроциты[17].
Комплементоопосредованный цитолиз клеток микроорганизмов осуществляет мембраноатакующий комплекс. Однако у большинства патогенов имеются толстые клеточные стенки или капсулы, которые не дают ему внедриться в их мембраны. Лишь несколько патогенных бактерий не развили у себя способность противостоять внедрению мембраноатакующего комплекса; в их числе бактерии рода Neisseria, имеющие очень тонкие стенки[17].
Связывась с комплексами антиген-антитело, белки комплемента повышают их растворимость и способствуют их разрушению фагоцитами. Накопление иммунных комплексов в кровотоке может приводить к их отложению на стенках сосудов и запуску воспаления, повреждающего сосуды и окружающие ткани. Белок C3d, образующийся в результате расщепления C3b, связывается с CR2 на поверхности B-клеток и способствует их активации и запуску гуморального иммунного ответа. C3d образуется в результате активации комплемента антигеном либо непосредственно, либо в комплексе с антителом. B-клетки могут связывать антиген посредством B-клеточных рецепторов и одновременно взаимодействовать с C3d посредством CR2, что приводит к усилению активирующего сигнала в B-клетках. Опсонизированные антигены также связываются фолликулярными дендритными клетками в герминальных центрах лимфоидных органов. Далее дендритные клетки представляют антиген B-клеткам в герминальном центре, что играет важную роль в отборе B-клеток, рецепторы которых обладают высоким сродством к антигену[18].
Противодействие патогенов
Патогены развили разнообразные механизмы, защищающие их от действия системы комплемента. Многие микроорганизмы имеют толстые клеточные стенки и капсулы, которые не дают мембраноатакующему комплексу встроиться в их клеточные мембраны. Такую относительно неспецифическую стратегию используют, в частности, грамположительные бактерии[19].
Микроорганизмы могут защищать себя от действия комплемента, заимствуя регуляторные белки комплемента организма-хозяина. Многие патогенные микроорганизмы несут на поверхности клеток большое количество сиаловой кислоты, которая привлекает фактор H, вытесняющий C3b из комплекса с Bb. Некоторые патогены, такие как шистосомы, Neisseria gonorrhoeae и некоторые виды рода Haemophilus[англ.], «крадут» остатки сиаловой кислоты у хозяина и присоединяют их к собственным полисахаридам. Другие, такие как Escherichia coli K1 и некоторые менингококки, имеют собственные биохимические пути синтеза сиаловой кислоты. У ряда патогенов имеются белки, привлекающие фактор H на их поверхность; такую стратегию используют бактерии Streptococcus pyogenes, Borrelia burgdorferi[англ.], N. gonorrhoeae, N. meningitidis, патогенные дрожжи Candida albicans и паразитические черви, такие как Echinococcus granulosus. Белок GP41[англ.] ВИЧ может связывать фактор H, что, как принято считать, защищает вирионы от разрушения. Кроме того, ВИЧ и ряд других возбудителей вставляют в свои липидные оболочки защитные белки хозяина, такие как DAF и CD59[20].
Некоторые патогены образуют специфические белки, которые мимикрируют под регуляторные белки комплемента. Так, E. coli экспрессирует C1q-связывающий белок, который не даёт C1q, C1r и C1s образовать комплекс. Staphylococcus aureus имеет белок SCIN, связывающий и стабильно подавляющий C3-конвертазы классического и альтернативного путей. Гликопротеин C-1 вируса простого герпеса дестабилизирует C3-конвертазу альтернативного пути, не давая ей взаимодействовать с пропердином. Мембранный белок GP160 паразита Trypanosoma cruzi связывает C3b и подавляет сборку C3-конвертазы. Вирус коровьей оспы имеет белок VCP-1, структурно близкий к C4BP. VCP-1 может взаимодействовать с C4b и C3b и способствует распаду C3- и C5-конвертаз[20].
Наконец, микроорганизмы могут подавлять развитие воспаления, вызванного активацией комплемента, при помощи специальных белков. Так, S. aureus экспрессирует белок CHIPS, который является антагонистом анафилотоксина C5a[20].
Эволюция
Хотя система комплемента изначально была описана у позвоночных животных, гомологи C3 и фактора B, а также примитивный вариант альтернативного пути найдены и у беспозвоночных. Белок C3, который разрезают и активируют сериновые протеазы, является родственником белка α2-макроглобулина[англ.], который является ингибитором сериновых протеаз и в ходе эволюции появился, вероятно, у общего предка современных позвоночных. Петля амплификации сигнала в альтернативном пути имеет древнее происхождение и имеется у иглокожих, у которых C3-конвертаза состоит из гомологов C3 и фактора B. Эти факторы экспрессируют амёбоидные целомоциты[англ.], которые циркулируют в целомической жидкости иглокожих. Экспрессия этих белков увеличивается при бактериальном заражении животного. Гомологи C3 беспозвоночных являются родственниками друг другу и формируют так называемое тиоэфирное белковое семейство (англ. thioester proteins, TEPs), получившее своё название по наличию характерной тиоэфирной связи у его членов. У комаров рода Anopheles образование белка TEP1[англ.] усиливается при инфекции, и TEP1 может непосредственно связываться с мембранами грамотрицательных бактерий, облегчая их фагоцитоз. Возможно, некоторые формы активности C3 появились ещё до возникновения двусторонне-симметричных животных, поскольку гены, родственные генам C3, фактора B и некоторых поздних компонентов комплемента имеются у коралловых полипов[21].
Эволюция системы комплемента, вероятно, происходила по пути возникновения новых путей активации. Самым первым, скорее всего, появился фиколиновый путь, который имеется у позвоночных и низших хордовых — оболочников. В геноме оболочника Ciona удалось выявить гены-гомологи MBL и C1q, а также двух сериновых протеаз из семейства MASP. Впоследствии, после появления адаптивного иммунитета и антител, у позвоночных возник классический путь активации, зависимый от антител[22].
Клиническое значение
Считается, что система комплемента может быть задействована в развитии ряда болезней, имеющих иммунную компоненту, таких как синдром Барракера — Симонса, бронхиальная астма, системная красная волчанка, гломерулонефрит, различные формы артрита, аутоиммунные заболевания сердца[англ.], рассеянный склероз, воспалительные заболевания кишечника, пароксизмальная ночная гемоглобинурия, атипичный гемолитико-уремический синдром, ишемически-реперфузионные повреждения[23][24] и отторжение пересаженных органов[25]. Продемонстрировано участие системы комплемента в развитии ряда заболеваний нервной системы, таких как болезнь Альцгеймера и другие нейродегенеративные расстройства, например, повреждения спинного мозга[26][27][28].
Недостаточность в работе терминальных стадий каскада комплемента служит предрасполагающим фактором в развитии аутоиммунных и инфекционных болезней, в частности, вызванных бактерией Neisseria meningitidis[29]. Инфекции, вызванные N. meningitidis и Neisseria gonorrhoeae, связаны с недостаточной работой мембраноатакующего комплекса (компоненты C5, C6, C7, C8, C9), который играет особую роль в защите от этих грамотрицательных бактерий[30][31]. 40—50 % пациентов, имеющих недостаточность по мембраноатакующему комплексу, страдают от возвратных инфекций, вызванных N. meningitidis[32].
Описаны мутации в генах, кодирующих C1q, C1r, C4, C2 и C3, причём недостаточная[англ.] активность C2 является наиболее частой формой недостаточности комплемента у людей. У более чем половины пациентов, имеющих мутации в C1q, C2 или C4, развивается системная красная волчанка, однако причины такой связи неизвестны. Возможно, описанная недостаточность приводит к неспособности эффективно удалять иммунные комплексы из сосудов, и оседание иммунных комплексов на стенках сосудов и в тканях вызывает хронические воспаления и аутоиммунные процессы. Кроме того, недостаточная работа комплемента не даёт эффективно разрушать апоптотические тельца, содержащие фрагментированную[англ.] ДНК, и именно апоптотические тельца, вероятно, являются главным источником ядерных антигенов, провоцирующих появление системной красной волчанки. Возможно также, что недостаточность по комплементу не даёт эффективно подавлять активность B-клеток, распознающих белки самого организма, что в итоге приводит к аутоиммунным заболеваниям. Недостаточность по компонентам C2 и C4 не всегда приводит к повышенной чувствительности к инфекциям, а нарушения работы C3 зачастую связаны с серьёзными, нередко фатальными, бактериальными заболеваниями[33].
Недостаточность по компонентам альтернативного пути активации комплемента, таким как фактор D и пропердин, приводит к повышенной предрасположенности к бактериальным инфекциям. Мутации, затрагивающие MBL, нередко связаны с иммунодефицитными состояниями[31].
Недостаточность по регуляторным белкам комплемента часто связана с ненормальной активацией комплемента. Недостаточность по ингибитору C1 наблюдается при аутосомно-доминантном заболевании, известном как наследственная ангионевротическая эдема[14]. Мутации, затрагивающие фактор H, являющийся регулятором системы комплемента, и мембранный кофактор CD46, связаны с развитием атипичного гемолитико-уремического синдрома[34][35]. Кроме того, распространённый однонуклеотидный полиморфизм гена, кодирующего фактор H, связан с распространённой возрастной макулодистрофией[36]. Полиморфизмы, затрагивающие компонент комплемента 3, фактор комплемента B и фактор комплемента I, также влияют на риск развития макулодистрофии[37]. Пароксизмальная ночная гемоглобинурия вызвана разрушением эритроцитов системой комплемента, которая происходит из-за отсутствия на эритроцитах GPI-заякоренных белков DAF и CD59[14], вызванного неспособностью синтезировать GPI-якорь[38]. Недостаточность по рецепторам комплемента CR3 и CR4, обусловленная мутациями в их одинаковой β-цепи, может приводить к недостаточности по адгезии лейкоцитов[31].
Многие патологические изменения, наблюдающиеся при бактериальных инфекциях, обусловлены не деятельностью бактерий напрямую, а острым воспалительным ответом, вызванным активацией комплемента. Иногда активация комплемента приводит к тромбозу, который может быть связан с ишемическими повреждениями тканей. Например, антитела, направленные против эндотелия сосудов пересаженного органа и иммунные комплексы, образовавшиеся в ходе аутоиммунного процесса, могут связываться с эндотелиальными клетками сосудов организма и активировать на них комплемент, что приводит к развитию воспаления и повреждениям сосудов. Кроме того, некоторые белки поздних стадий каскада комплемента могут напрямую активировать протромбиназы. Иммунные комплексы могут откладываться также на стенках почечных канальцев, что приводит к гломерулонефриту[31].
К числу диагностических методов, оценивающих работу системы комплемента, относят тест на общую активность комплемента[англ.][39].
История изучения

Система комплемента стала первой известной гуморальной системой врождённого иммунитета. В 1888 году Джордж Генри Наттолл обнаружил, что сыворотка овечьей крови обладает умеренным действием против бактерии, вызывающей сибирскую язву, причём это свойство сыворотки исчезает после её нагревания[40]. В 1891 году Ганс Эрнст Август Бухнер описал те же свойства сыворотки крови по отношению к микроорганизмам, и назвал эту особенность «алексин»[41]. К 1894 году в нескольких лабораториях было показано, что сыворотка крови морских свинок, переболевших холерой, убивает холерные вибрионы in vitro, причём защитные свойства сыворотки исчезают после теплового воздействия. В 1898 году Жюль Борде, сотрудник Института Пастера (Париж), изучал иммунный гемолиз и описал термолабильную составляющую системы факторов, ответственных за этот процесс. Позднее Пауль Эрлих предложил называть фракцию, описанную Борде, словом «комплемент» от лат. complementare — дополнять. Последующие открытия показали, что комплемент представляет собой не один белковый фактор, а сложную белковую систему. В 50-х годах XX века Л. Пиллемер описал пропердиновую систему, которую в 70-х годах было предложено называть альтернативным путём активации системы комплемента, а антителозависимый путь активации, описанный Ж. Борде, стали называть классическим. В 90-х годах XX века получил признание третий путь активации системы комплемента — лектиновый[5].
Примечания
- ↑ 1 2 Abbas, Lichtman, Pillai, 2015, p. 272.
- ↑ Галактионов, 2004, с. 287.
- ↑ Murphy, Weaver, 2017, p. 50.
- ↑ Abbas, Lichtman, Pillai, 2015, p. 272—273.
- ↑ 1 2 Ярилин, 2010, с. 167.
- ↑ Abbas, Lichtman, Pillai, 2015, p. 276—278.
- ↑ 1 2 Abbas, Lichtman, Pillai, 2015, p. 278.
- ↑ Abbas, Lichtman, Pillai, 2015, p. 273—276.
- ↑ Abbas, Lichtman, Pillai, 2015, p. 279—280.
- ↑ 1 2 3 Abbas, Lichtman, Pillai, 2015, p. 280.
- ↑ Abbas, Lichtman, Pillai, 2015, p. 280—281.
- ↑ 1 2 3 Abbas, Lichtman, Pillai, 2015, p. 281.
- ↑ Abbas, Lichtman, Pillai, 2015, p. 281—282.
- ↑ 1 2 3 Abbas, Lichtman, Pillai, 2015, p. 282.
- ↑ Abbas, Lichtman, Pillai, 2015, p. 282—283.
- ↑ Abbas, Lichtman, Pillai, 2015, p. 283—284.
- ↑ 1 2 3 4 5 Abbas, Lichtman, Pillai, 2015, p. 284.
- ↑ Abbas, Lichtman, Pillai, 2015, p. 284—285.
- ↑ Abbas, Lichtman, Pillai, 2015, p. 286—287.
- ↑ 1 2 3 Abbas, Lichtman, Pillai, 2015, p. 287.
- ↑ Murphy, Weaver, 2017, p. 61—62.
- ↑ Murphy, Weaver, 2017, p. 62.
- ↑ Arumugam T. V., Shiels I. A., Woodruff T. M., Granger D. N., Taylor S. M. The role of the complement system in ischemia-reperfusion injury. (англ.) // Shock (Augusta, Ga.). — 2004. — May (vol. 21, no. 5). — P. 401—409. — doi:10.1097/00024382-200405000-00002. — PMID 15087815.
- ↑ Naesens M., Li L., Ying L., Sansanwal P., Sigdel T. K., Hsieh S. C., Kambham N., Lerut E., Salvatierra O., Butte A. J., Sarwal M. M. Expression of complement components differs between kidney allografts from living and deceased donors. (англ.) // Journal Of The American Society Of Nephrology : JASN. — 2009. — August (vol. 20, no. 8). — P. 1839—1851. — doi:10.1681/ASN.2008111145. — PMID 19443638.
- ↑ Sacks S. H., Chowdhury P., Zhou W. Role of the complement system in rejection. (англ.) // Current Opinion In Immunology. — 2003. — October (vol. 15, no. 5). — P. 487—492. — doi:10.1016/s0952-7915(03)00100-6. — PMID 14499254.
- ↑ Galvan M. D., Luchetti S., Burgos A. M., Nguyen H. X., Hooshmand M. J., Hamers F. P., Anderson A. J. Deficiency in complement C1q improves histological and functional locomotor outcome after spinal cord injury. (англ.) // The Journal Of Neuroscience : The Official Journal Of The Society For Neuroscience. — 2008. — 17 December (vol. 28, no. 51). — P. 13876—13888. — doi:10.1523/JNEUROSCI.2823-08.2008. — PMID 19091977.
- ↑ Nguyen H. X., Galvan M. D., Anderson A. J. Characterization of early and terminal complement proteins associated with polymorphonuclear leukocytes in vitro and in vivo after spinal cord injury. (англ.) // Journal Of Neuroinflammation. — 2008. — 25 June (vol. 5). — P. 26—26. — doi:10.1186/1742-2094-5-26. — PMID 18578885.
- ↑ Beck K. D., Nguyen H. X., Galvan M. D., Salazar D. L., Woodruff T. M., Anderson A. J. Quantitative analysis of cellular inflammation after traumatic spinal cord injury: evidence for a multiphasic inflammatory response in the acute to chronic environment. (англ.) // Brain : A Journal Of Neurology. — 2010. — February (vol. 133, no. Pt 2). — P. 433—447. — doi:10.1093/brain/awp322. — PMID 20085927.
- ↑ Brown E. J. Interaction of Gram-Positive Microorganisms with Complement (англ.) // Current Topics in Microbiology and Immunology. — 1985. — P. 159—187. — ISBN 9783642456060. — ISSN 0070-217X. — doi:10.1007/978-3-642-45604-6_8.
- ↑ Ram S., Lewis L. A., Rice P. A. Infections of people with complement deficiencies and patients who have undergone splenectomy. (англ.) // Clinical Microbiology Reviews. — 2010. — October (vol. 23, no. 4). — P. 740—780. — doi:10.1128/CMR.00048-09. — PMID 20930072.
- ↑ 1 2 3 4 Abbas, Lichtman, Pillai, 2015, p. 286.
- ↑ Lewis L. A., Ram S. Meningococcal disease and the complement system. (англ.) // Virulence. — 2014. — 1 January (vol. 5, no. 1). — P. 98—126. — doi:10.4161/viru.26515. — PMID 24104403.
- ↑ Abbas, Lichtman, Pillai, 2015, p. 285—286.
- ↑ Dragon-Durey M. A., Frémeaux-Bacchi V. Atypical haemolytic uraemic syndrome and mutations in complement regulator genes. (англ.) // Springer Seminars In Immunopathology. — 2005. — November (vol. 27, no. 3). — P. 359—374. — doi:10.1007/s00281-005-0003-2. — PMID 16189652.
- ↑ Zipfel P. F., Misselwitz J., Licht C., Skerka C. The role of defective complement control in hemolytic uremic syndrome. (англ.) // Seminars In Thrombosis And Hemostasis. — 2006. — March (vol. 32, no. 2). — P. 146—154. — doi:10.1055/s-2006-939770. — PMID 16575689.
- ↑ Mooijaart S. P., Koeijvoets K. M., Sijbrands E. J., Daha M. R., Westendorp R. G. Complement Factor H polymorphism Y402H associates with inflammation, visual acuity, and cardiovascular mortality in the elderly population at large. (англ.) // Experimental Gerontology. — 2007. — November (vol. 42, no. 11). — P. 1116—1122. — doi:10.1016/j.exger.2007.08.001. — PMID 17869048.
- ↑ Bradley D. T., Zipfel P. F., Hughes A. E. Complement in age-related macular degeneration: a focus on function. (англ.) // Eye (London, England). — 2011. — June (vol. 25, no. 6). — P. 683—693. — doi:10.1038/eye.2011.37. — PMID 21394116.
- ↑ Parker C., Omine M., Richards S., Nishimura J., Bessler M., Ware R., Hillmen P., Luzzatto L., Young N., Kinoshita T., Rosse W., Socié G., International PNH Interest Group. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. (англ.) // Blood. — 2005. — 1 December (vol. 106, no. 12). — P. 3699—3709. — doi:10.1182/blood-2005-04-1717. — PMID 16051736.
- ↑ Complement Deficiencies Workup: Laboratory Studies, Imaging Studies, Other Tests (англ.). emedicine.medscape.com. Дата обращения: 26 апреля 2018. Архивировано 27 апреля 2018 года.
- ↑ Chaplin Jr. H. Review: the burgeoning history of the complement system 1888-2005. (англ.) // Immunohematology. — 2005. — Vol. 21, no. 3. — P. 85—93. — PMID 16178664.
- ↑ Nesargikar P. N., Spiller B., Chavez R. The complement system: history, pathways, cascade and inhibitors. (англ.) // European Journal Of Microbiology & Immunology. — 2012. — June (vol. 2, no. 2). — P. 103—111. — doi:10.1556/EuJMI.2.2012.2.2. — PMID 24672678.
Литература
- Галактионов В. Г. Иммунология. — М. : Издат. центр «Академия», 2004. — 528 с. — ISBN 5-7695-1260-1.
- Песнякевич А. Г. 1.1.5. Система комплемента // Иммунология : учеб. пособие. — Минск : БГУ, 2018. — С. 43—55. — 255 с. — УДК 28.074я73 П28(G). — ISBN 978-985-566-628-9.
- Ярилин А. А. Иммунология. — М. : ГЭОТАР-Медиа, 2010. — 752 с. — ISBN 978-5-9704-1319-7.
- Abul K. Abbas, Andrew H. Lichtman, Shiv Pillai. Cellular and Molecular Immunology. — Philadelphia : Elsevier Saunders, 2015. — ISBN 978-0-323-22275-4.
- Kenneth Murphy, Casey Weaver. Janeway's Immunobiology. — Garland Science, 2017. — ISBN 978-0-8153-4505-3.
| В библиографических каталогах |
|---|