Тяжёлый комбинированный иммунодефицит
| Тяжёлый комбинированный иммунодефицит | |
|---|---|
| МКБ-11 | 4A01.10 |
| МКБ-10 | D81.0, D81.2 и D81.1 |
| МКБ-9 | 279.2 |
| DiseasesDB | 11978 |
| MeSH | D016511 |
Тяжёлый комбинированный иммунодефицит, (англ. SCID, также алимфоцитоз, синдром Глянцмана-Риникера, синдром тяжёлого комбинированного иммунодефицита, и тимическая алимфоплазия[1]) — это генетическое заболевание, при котором в результате дефекта одного из генов нарушается работа компонентов адаптивной иммунной системы B- и T-лимфоцитов. Тяжёлый комбинированный иммунодефицит— это тяжёлая форма наследственного иммунодефицита, который также известен как синдром мальчика в пузыре, так как больные крайне уязвимы перед инфекционными болезнями и вынуждены находиться в стерильной среде. Одним из таких больных был Дэвид Веттер. Тяжёлый комбинированный иммунодефицит является результатом настолько сильного повреждения иммунной системы, что последняя считается практически отсутствующей.
Симптомами тяжёлого комбинированного иммунодефицита могут являться хроническая диарея, ушные инфекции, возвратный пневмоцистоз, обильные кандидозы полости рта. Без лечения, в случае, если не было произведено успешной трансплантации гемопоэтических стволовых клеток, дети обычно умирают в течение первого года жизни от тяжёлых рецидивирующих инфекций.
Распространённость
Наиболее часто цитируемый показатель распространённости тяжёлого комбинированного иммунодефицита составляет примерно 1 на 100,000 родившихся, хотя некоторыми такие данные рассматриваются как недооценка истинной распространённости[2]. В Австралии сообщается о такой частоте встречаемости, как 1 на 65,000 родившихся[3].
Недавно проведённые исследования показали, что в популяции навахо 1 ребёнок из каждых 2500 наследует тяжёлый комбинированный иммунодефицит. Это является причиной значительного процента заболеваемости и смертности среди детей данной народности[4]. Текущие исследования выявили аналогичную картину у племён Апачи[5].
Типы
| Тип | Описание |
|---|---|
| X-сцепленный тяжёлый иммунодефицит | Наиболее распространённый тип тяжёлого комбинированного иммунодефицита, возникающий из-за мутаций в гене, кодирующем общие гамма-цепи, белок которых является общим для рецепторов интерлейкинов IL-2, IL-4, IL-7, IL-9, IL-15 и IL-21. Перечисленные интерлейкины и их рецепторы вовлечены в процессы развития T- и B-лимфоцитов. В результате мутаций происходят дисфункции общей гамма-цепи, и, как следствие, дефект распространяется на процесс сигнализации интерлейкина. Происходит почти полный отказ иммунной системы как со стороны развития, так и со стороны функционирования, с отсутствием или очень малым количеством T-лимфоцитов, NK-клеток и нефункциональными B-лимфоцитами. Общая гамма-цепь кодируется геном IL-2 рецепторов гамма, который находится на X-хромосоме. Наследуется как рецессивный признак. |
| Дефицит аденозиндеаминазы | Второй по распространённости тип тяжёлого комбинированного иммунодефицита. Его причиной является дефект фермента аденозиндеамиазы, который необходим для расщепления пуринов. Недостаток аденозиндеаминазы провоцирует накопление dATP. Этот метаболит ингибирует активность фермента рибонуклеотидредуктазы, участвующего в превращении рибонуклеотидов в дезоксирибонуклеотиды. Эффективность иммунной системы зависит от пролиферации лимфоцитов и, следовательно, синтеза dNTP. Если рибонуклеотидредуктаза не способна нормально функционировать, пролиферация лимфоцитов блокируется, а иммунная система компрометируется. |
| Синдром Оменна | Производство иммуноглобулинов требует участия рекомбинантного фермента, полученного от рекомбинации генов, активирующих RAG-1 и RAG-2. Эти ферменты участвуют в первом этапе V(D)J рекомбинации, в котором сегменты B-лимфоцитов или ДНК T-лимфоцитов перестраиваются, создавая новые T- или B-клеточный рецепторы. Некоторые мутации RAG-1 или RAG-2 продотвращают процесс V(D)J рекомбинации, тем самым приводя к возникновения ТКТД[6]. |
| Синдром голых лимфоцитов | MHC класса II не экспрессируется на поверхности антигенпредставляющих клеток. Аутосомно-рецессивный тип наследования. |
| Дефицит JAK3 | JAK3 является ферментом, который выступает посредником трансдукции через общую гамма-цепь. Мутация гена JAK3 также вызывает тяжёлый комбинированный иммунодефицит[7]. |
| Дефицит DCLRE1C/Artemis | Несмотря на то, что исследователями было идентифицировано около дюжины генов, вызывающих ТКИД, население Навахо и Апачи страдает наиболее тяжёлой формой заболевания. Это связано с отсутствием гена DCLRE1C/Artemis. Без этого гена организм ребёнка не в состоянии восстановить ДНК или вырабатывать антитела. |
Диагностика
В нескольких штатах США проводятся экспериментальные исследования для диагностики тяжёлого комбинированного иммунодефицита новорождённых при помощи иссечения рекомбинантных T-лимфоцитов. По состоянию на 1 февраля 2009 года, В Висконсине и Массачусетсе проводится скрининг новорожденных на предмет выявления этой патологии[8][9]. В Мичигане скрининг на тяжёлый комбинированный иммунодефицит начали в октябре 2011 года[10]. Однако стандартизированное тестирование этого заболевания в настоящее время недоступно в связи с разнообразием генетического дефекта у новорожденных. Некоторые формы тяжёлого комбинированного иммунодефицита могут быть обнаружены путём секвенирования ДНК плода, если есть основания подозревать данное заболевание. В противном случае, наследственное заболевание не диагностируется примерно до 6 месяцев. Как правило, на его наличие могут указывать рецидивирующие инфекции. Задержка в обнаружении тяжёлого комбинированного иммунодефицита обусловлена тем, что у новорожденных в течение первых нескольких недель жизни присутствуют антитела матери, и дети с таким иммунодефицитом выглядят здоровыми.
Лечение
Наиболее распространённым методом лечения тяжёлого комбинированного иммунодефицита является трансплантация гемопоэтических стволовых клеток, которая проходит успешно либо при участии неродственного донора, либо при участии полу-совместимого донора, которым может являться один из родителей. Последний вид трансплантации носит название «гаплоидентичной» и был усовершенствован в Мемориальном онкологическом центре им. Слоуна-Кеттеринга в Нью-Йорке, а также в Медицинском центре дьюкского университета, где в настоящее время проведено наибольшее количество подобных пересадок[11]. При гаплоидентичной пересадке костного мозга необходимо наличие донорского костного мозга, чтобы избежать гомологичной реакции при использовании всех зрелых T-клеток[12]. Следовательно, функциональность иммунной системы развивается дольше у пациента, получающего костный мозг. Дэвид Веттер, один из первых, кому была проведена подобная операция, в итоге умер от вируса Эпштейна — Барр, которым был заражён костный мозг, пересаженный от его сестры. Сегодня пересадка, сделанная в первые три месяца жизни ребёнка, имеет высокий уровень успешности. Также врачи успешно проводили внутриутробную трансплантацию, сделанную до рождения ребёнка, с использованием пуповинной крови, богатой стволовыми клетками. Внутриутробная трансплантация позволяет иммунной системе плода развиваться в стерильной среде матки[13]. Однако такое осложнение, как гомологичная болезнь, довольно сложно обнаружить[14].
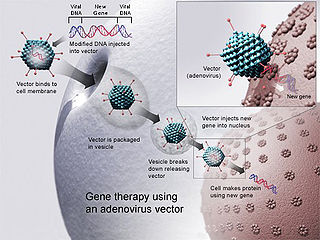
В качестве альтернативы пересадке костного мозга была предложена генотерапия. В 1990 году 4-летняя Ашанти де Сильва стала первой пациенткой, которая успешно прошла курс генной терапии. Исследователи собрали образцы крови Ашанти, изолировали некоторые лимфоциты, а затем использовали вирус, чтобы вставить в геном гены аденозиндезаминазы дикого типа. Затем эти клетки вводили обратно в организм и они начинали синтезировать нормальный фермент. Дефицит аденозиндезаминазы компенсировался дополнительными еженедельными инъекциями[15]. Тем не менее в 2000 году испытания были приостановлены, поскольку обнаружилось, что 2 из 10 пациентов в результате генотерапии заболели лейкозом в результате введения гена, несущего ретровирус, возле онкогена. Позднее метод был модифицирован благодаря чему из 10 детей, рожденных с этим смертельным иммунным расстройством и прошедших экспериментальную терапию в период с 2009 по 2012 год, 9 остаются здоровыми (по данным на 2021 год). Эта терапия наиболее эффективна у детей младшего возраста, поскольку старший ребенок, которому в то время было 15 лет, был единственным участником, чья иммунная функция не была восстановлена лечением генотерапией[16][17]. С тех пор исследователи перешли к использованию модифицированных лентивирусов в качестве вирусного вектора для генной терапии. Эти вирусы могут проникать в ядра неделящихся клеток, что означает, что они должны быть более безопасными и эффективными в контексте генной терапии. Согласно опубликованным результатам испытания генной терапии ADA-SCID с использованием лентивирусного вектора в фазе 1/2, 48 из 50 детей, получавших генную терапию, все еще были практически излечены от болезни в течение трехлетнего периода наблюдения[16][17].
На сентябрь 2020 года в процессе исследования и доведения до клинических испытаний находится новый потенциальный способ восстановления функциональной иммунной системы младенцев с тяжелым комбинированным иммунодефицитом с использованием технологии редактирования генома CRISPR, предложенный Израильской исследовательской лабораторией[18].
Есть также некоторые нелечебные методы терапии тяжёлого комбинированного иммунодефицита. Обратная изоляция предполагает использование ламинарного потока воздуха и механических барьеров (для избежания физического контакта с другими людьми), чтобы изолировать пациента от любых вредных патогенных микроорганизмов, присутствующих во внешней среде[19].
Примечания
- ↑ Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. ISBN 1-4160-2999-0
- ↑ NEWBORN SCREENING FOR PRIMARY IMMUNODEFICIENCY DISEASE. Дата обращения: 21 сентября 2012. Архивировано 22 августа 2016 года.
- ↑ Yee A, De Ravin SS, Elliott E, Ziegler JB (2008). «Severe combined immunodeficiency: A national surveillance study». Pediatr Allergy Immunol 19 (4): 298—302. doi:10.1111/j.1399-3038.2007.00646.x. PMID 18221464
- ↑ a b «News From Indian Country — A rare and once-baffling disease forces Navajo parents to cope». Retrieved 2008-03-01
- ↑ a b Li L, Moshous D, Zhou Y et al. (2002). «A founder mutation in Artemis, an SNM1-like protein, causes SCID in Athabascan-speaking Native Americans». J. Immunol. 168 (12): 6323-9. PMID 12055248
- ↑ Haq IJ, Steinberg LJ, Hoenig M et al. (2007). «GvHD-associated cytokine polymorphisms do not associate with Omenn syndrome rather than T-B- SCID in patients with defects in RAG genes». Clin. Immunol. 124 (2): 165-9. doi:10.1016/j.clim.2007.04.013. PMID 17572155
- ↑ Pesu M, Candotti F, Husa M, Hofmann SR, Notarangelo LD, O’Shea JJ (2005). «Jak3, severe combined immunodeficiency, and a new class of immunosuppressive drugs». Immunol. Rev. 203: 127-42. doi:10.1111/j.0105-2896.2005.00220.x. PMID 15661026
- ↑ «Wisconsin First State in Nation to Screen All Newborns for Severe Combined Immune Deficiency (SCID) or „Bubble Boy Disease“»
- ↑ «NEWBORN SCREENING FOR PRIMARY IMMUNODEFICIENCY DISEASE»
- ↑ «MDCH Adds Severe Combined Immunodeficiency (SCID) to Newborn Screening»
- ↑ «Severe Combined Immunodeficiency (SCID): Immunodeficiency Disorders: Merck Manual Professional». Retrieved 2008-03-01
- ↑ a b Chinen J, Buckley RH (2010). «Transplantation immunology: solid organ and bone marrow». J. Allergy Clin. Immunol. 125 (2 Suppl 2): S324-35
- ↑ Vickers, Peter S. (2009). Severe combined immune deficiency: early hospitalisation and isolation. Hoboken NJ: John Wiley & Sons, 29-47. ISBN 978-0-470-74557-1
- ↑ Buckley RH (2004). «Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution». Annu. Rev. Immunol. 22 (1): 625—655
- ↑ a b Fischer A, Hacein-Bey S, Cavazzana-Calvo M (2002). «Gene therapy of severe combined immunodeficiencies». Nat Rev Immunol 2 (8): 615—621
- ↑ 1 2 Williams S.C.P. (2021). A decade after gene therapy, children born with deadly immune disorder remain healthy Архивная копия от 21 октября 2021 на Wayback Machine. UCLA researchers provide update on patients treated for ADA-SCID between 2009 and 2012. UCLA Newsroom
- ↑ 1 2 Reinhardt, B., Habib, O., Shaw, K. L., Garabedian, E., Carbonaro-Sarracino, D. A., Terrazas, D., ... & Kohn, D. B. (2021). Long-term Outcomes after Gene Therapy for Adenosine Deaminase Severe Combined Immune Deficiency (ADA SCID). Blood. 138(15), 1304-1316 PMID 33974038 doi:10.1182/blood.2020010260
- ↑ Interview: Israeli researchers are developing a CRISPR gene editing therapy for SCID, interview with Ayal Hendel - CRISPR Medicine. Дата обращения: 4 сентября 2020. Архивировано 19 сентября 2020 года.
- ↑ Tamaroff MH, Nir Y, Straker N (1986). «Children reared in a reverse isolation environment: effects on cognitive and emotional development». J. Autism Dev. Disord. 16 (4): 415—424