Фенилкетонурия
| Фенилкетонурия | |
|---|---|
 Фенилаланин | |
| МКБ-11 | 5C50.0 |
| МКБ-10 | E70.0 |
| МКБ-9 | 270.1 |
| МКБ-9-КМ | 270.1[1] |
| OMIM | 261600 |
| DiseasesDB | 9987 |
| MedlinePlus | 001166 |
| eMedicine | ped/1787 derm/712 |
| MeSH | D010661 |
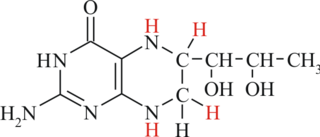
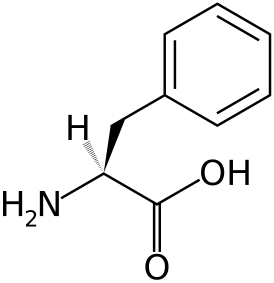
Фенилкетонури́я — наследственное заболевание группы ферментопатий, связанное с нарушением метаболизма аминокислот, главным образом фенилаланина. Несоблюдение низкобелковой диеты сопровождается накоплением фенилаланина и его токсических продуктов, что приводит к тяжёлому поражению ЦНС, проявляющемуся, в частности, в виде нарушения умственного развития (фенилпировиноградной олигофрении). Одно из немногих наследственных заболеваний, поддающихся успешному лечению.
История
Открытие фенилкетонурии связывают с именем норвежского врача Ивара Асбьёрна Фёллинга, описавшего в 1934 году гиперфенилаланинемию, ассоциированную с задержкой умственного развития[2]. В Норвегии заболевание также известно под названием «болезни Фёллинга» (норв. Føllings sykdom) в честь открывателя.
Успешное лечение впервые было разработано и проведено в Англии (Бирмингемский детский госпиталь) группой медицинских работников под руководством Хорста Биккеля в начале 50-х годов XX века, однако настоящий успех пришёл только после широкого применения ранней диагностики фенилкетонурии по повышенному содержанию фенилаланина в крови у новорождённых (метод Гатри, разработанный и внедренный в 1958—1961 гг).
С развитием генетики и накоплением опыта в диагностике и лечении фенилкетонурии стало ясно, что за это заболевание «отвечает» единственный ген, называемый PAH[англ.][3] (12q23.2[4]; ген фенилаланингидроксилазы[5]).
Выделены и описаны атипичные формы фенилкетонурии, разработаны новые методы лечения, в ближайшей перспективе — генотерапия этого тяжёлого заболевания, ставшего классическим образцом успешного оказания медицинской и организационной помощи при наследственной патологии.
Этиология
В большинстве случаев заболевание связано с резким снижением или полным отсутствием активности печёночного фермента фенилаланин-4-гидроксилазы, который в норме катализирует превращение фенилаланина в тирозин. До 1 % случаев фенилкетонурии представлено атипичными формами, связанными с мутациями в других генах, отвечающих за кодирование ферментов, обеспечивающих синтез кофактора фенилаланингидроксилазы — тетрагидробиоптерина (BH4). Заболевание наследуется по аутосомно-рецессивному типу.
Распространённость заболевания варьирует в различных группах населения. Например, среди европеоидных жителей США встречается в среднем в 1 случае на 10 000.[6] Наиболее высокий уровень — в Турции: 1 из 2600. В Финляндии и Японии уровень фенилкетонурии крайне низок: менее 1 случая на 119 000 рождений. В исследовании 1987 года в Словакии среди отдельных цыганских популяций были обнаружены сверхвысокие уровни фенилкетонурии из-за инбридинга: 1 случай на 40 рождений.[7] Мужчины и женщины страдают в равной степени[8].
| Страна | Встречаемость заболевания |
|---|---|
| Китай | 1 на 18000[9] |
| Финляндия | менее 1 на 100000[10] |
| Ирландия | 1 на 4500[11] |
| Япония | 1 на 120000[12] |
| Корея | 1 на 41000[13] |
| Норвегия | 1 на 13000[14] |
| Турция | 1 на 2600 |
| Индия | 1 на 18300 |
| США | 1 на 15000[15] |
Патогенез
Вследствие метаболического блока активируются побочные пути обмена фенилаланина, и в организме происходит накопление его токсичных производных — фенилпировиноградной и фенилмолочной кислот, которые в норме практически не образуются. Кроме того, образуются также почти полностью отсутствующие в норме фенилэтиламин и ортофенилацетат, избыток которых вызывает нарушение метаболизма липидов в головном мозге. Предположительно, это и ведёт к прогрессирующему снижению интеллекта у таких больных вплоть до идиотии. Окончательно механизм развития нарушений функций мозга при фенилкетонурии остаётся неясным. Среди причин также предполагается дефицит нейромедиаторов мозга, вызванный относительным снижением количества тирозина и других «больших» аминокислот, конкурирующих с фенилаланином при переносе через гемато-энцефалический барьер, и прямое токсическое действие фенилаланина.
Диагностика
Производится полуколичественным тестом или количественным определением фенилаланина в крови. При нелеченных случаях возможно выявление продуктов распада фенилаланина (фенилкетонов) в моче (не ранее 10—12 дня жизни ребёнка). Также возможно определение активности фермента фенилаланингидроксилазы в биоптате печени и поиск мутаций в гене фенилаланингидроксилазы. Для диагностики 2 и 3 типа, связанных с мутацией в гене, отвечающем за синтез кофактора, необходимы дополнительные диагностические исследования.
В возрасте от 2—4 месяцев у больных появляются такие симптомы, как вялость, судороги, гиперрефлексия, «мышиный» запах пота и мочи[16] или «запах волка»[17], экзема. Также среди других симптомов отмечены: мышечная гипертензия, гиперкинезы, неустойчивая походка, при несоблюдении диеты светлеют глаза, волосы, кожа (по причине недостаточного количества в организме меланина, производного тирозина); судорожные припадки[18].
Психическое состояние
Поздно выявленная, а также нелеченная или пропущенная фенилкетонурия может сопровождаться глубокой степенью умственной отсталости, обычно идиотией или имбецильностью[18], могут наблюдаться явления эхопраксии (повторение движений окружающих) и эхолалии (повторение речи)[18], также характерна вялость с редкими вспышками злобы и раздражительности[18].
Лечение и профилактика
При своевременной диагностике патологических изменений можно полностью избежать, если с рождения и на всю жизнь ограничить поступление в организм фенилаланина с пищей.
Позднее начало лечения хотя и даёт определённый эффект, но не устраняет развившихся ранее необратимых изменений тканей мозга.
Некоторые из современных газированных напитков, жевательных резинок и лекарственных препаратов содержат фенилаланин в форме дипептида (аспартам), о чём производители обязаны предупреждать на этикетке. Так, например, на этикетках ряда безалкогольных напитков после указания состава и пищевой ценности 100 мл напитка приводится следующее предупреждение: «Содержит источник фенилаланина. Противопоказано применение при фенилкетонурии».
При рождении ребёнка в роддомах на 3—4 сутки берут анализ крови и проводят неонатальный скрининг для обнаружения врождённых заболеваний обмена веществ. На этом этапе возможно обнаружение фенилкетонурии, и, как следствие, возможно раннее начало лечения для предотвращения необратимых последствий.
В Российской Федерации детям в возрасте до 18 лет по этому заболеванию безусловно устанавливается категория «ребёнок-инвалид»[19].
Лечение проводится в виде строгой диеты от обнаружения заболевания как минимум до полового созревания, многие авторы придерживаются мнения о необходимости пожизненной диеты. Диета исключает мясные, рыбные, молочные продукты и другие продукты, содержащие животный и, частично, растительный белок. Дефицит белка восполняется аминокислотными смесями без фенилаланина. Кормление грудью детей, больных фенилкетонурией, возможно и может быть успешным при соблюдении некоторых ограничений[20][21].
Расчет диеты для больного фенилкетонурией проводит врач с учётом потребности в фенилаланине и его допустимом количестве.
Допустимое количество фенилаланина для больных фенилкетонурией[22]
| Возраст детей | Суточное количество фенилаланина (мг/кг массы тела) |
|---|---|
| До 2 мес. | 60 |
| 2—3 мес. | 60—55 |
| 3—6 мес. | 55—45 |
| 6—12 мес. | 45—35 |
| 1—1,5 года | 35—30 |
| 1,5—3 года | 30—25 |
| 3—6 лет | 25—15 |
| Старше 6 лет | 15—10 |
Некоторые (мягкие) формы заболевания поддаются лечению кофактором (тетрагидробиоптерином) поражённого фермента (фенилаланингидроксилазы). Разрабатываются новые подходы к лечению фенилкетонурии — использование заместительной терапии фенилаланинлиазой (PAL, пегвалиаза) — растительным ферментом, превращающим фенилаланин в безвредные метаболиты, и генотерапия на основе введения в организм вирусного вектора, содержащего ген фенилаланингидроксилазы. Эти методы пока не вышли из стен лабораторий. Атипичные формы в меньшей степени поддаются диетотерапии и лечатся введением препаратов тетрагидробиоптерина или его синтетических аналогов (сапроптерин).[23]
См. также
- Недостаточность тетрагидробиоптерина — группа заболеваний, некоторые из которых сопровождаются повышением уровней фенилаланина.
- Слабовыраженная гиперфенилаланинемия без дефицита тетрагидробиоптерина — родственное заболевание, открытое в 2017 году.
Примечание
- Сайт Латыпова с научной и популярной информацией по фенилкетонурии
- Фенилкетонурия: причины, симптомы, лечение // Журнал «9 Месяцев», № 04, 2004
- Информационный интернет-журнал и форум для родителей детей больных фенилкетонурией, 2007—2011
- Сайт московской организации помощи больным фенилкетонурией («Общество пациентов с фенилкетонурией»), 2014
- Сайт Саратовского объединения помощи людям с фенилкетонурией
Примечания
- ↑ Disease Ontology (англ.) — 2016.
- ↑ Folling, A. Ueber Ausscheidung von Phenylbrenztraubensaeure in den Harn als Stoffwechselanomalie in Verbindung mit Imbezillitaet (нем.) // Ztschr. Physiol. Chem. : magazin. — 1934. — Bd. 227. — S. 169—176.
- ↑ Heidi Chial. Rare Genetic Disorders: Learning About Genetic Disease Through Gene Mapping, SNPs, and Microarray Data // Nature Education. — 2008. — № 1. — С. 192. Архивировано 7 апреля 2015 года.
- ↑ PHENYLKETONURIA; PKU Архивная копия от 29 мая 2014 на Wayback Machine // Online Mendelian Inheritance in Man
- ↑ Ген фенилаланингидроксилазы: мутации и фенилкетонурия. Дата обращения: 8 января 2016. Архивировано 27 января 2016 года.
- ↑ Bickel, H.;, Bachmann, C.; Beckers, R.; Brandt, N.J.; Clayton, B.E.; Corrado, G; et al. Neonatal mass screening for metabolic disorders: summary of recent sessions of the committee of experts to study inborn metabolic diseases (англ.) // public health committee, Eur. J. Pediatr. : journal. — 1981. — P. 133—139. — doi:10.1007/BF00441305.
- ↑ Ferák, V.;, Siváková, D.; Sieglová, Z. Slovenskí Cigáni (Rómovia) – populácia s najvyšším koeficientom inbrídingu v Európe. (словац.) // Bratislavské lekárske listy (Bratislava Medical Journal). — 1987. — Zv. 87. — S. 168—175.
- ↑ Karen Marcdante, Robert M. Kliegman. Nelson Essentials of Pediatrics E-Book. — Elsevier Health Sciences, 2014-02-25. — 779 с. — ISBN 9780323226981. Архивировано 11 сентября 2017 года.
- ↑ Liu, S.R.; Zuo, Q.H. Newborn screening for phenylketonuria in eleven districts (англ.) // Chinese Medical Journal[англ.] : journal. — 1986. — Vol. 99. — P. 113—118.
- ↑ Guldberg, P., Henriksen, K. F., Sipila, I., Guttler, F., de la Chapelle, A. Phenylketonuria in a low incidence population: molecular characterization of mutations in Finland (англ.) // Journal of Medical Genetics[англ.] : journal. — 1995. — Vol. 32, no. 12. — P. 976—978. — doi:10.1136/jmg.32.12.976. — PMID 8825928. — PMC 1051781.
- ↑ DiLella, A. G., Kwok, S. C. M., Ledley, F. D., Marvit, J., Woo, S. L. C. Molecular structure and polymorphic map of the human phenylalanine hydroxylase gene (англ.) // Biochemistry : journal. — 1986. — Vol. 25, no. 4. — P. 743—749. — doi:10.1021/bi00352a001. — PMID 3008810.
- ↑ Aoki, K.; Wada, Y. Outcome of the patients detected by newborn screening in Japan (англ.) // Acta Paediatr. Jpn. : journal. — 1988. — Vol. 30, no. 4. — P. 429—434. — doi:10.1111/j.1442-200X.1988.tb02533.x. — PMID 3150232.
- ↑ Lee, D.H.; Koo, S.K.; Lee, K.S.; Yeon, Y.J.; Oh, H.J.; Kim, S.W.;Lee, S.J. ; Kim, S.S.; Lee, J.E.; Jo, I.; Jung, S.C. The molecular basis of phenylketonuria in Koreans (англ.) // Journal of Human Genetics[англ.]. — 2004. — Vol. 49, no. 1. — P. 617—621. — doi:10.1007/s10038-004-0197-5. — PMID 15503242.
- ↑ Oslo universitetssykehus. Дата обращения: 27 июня 2021. Архивировано из оригинала 1 апреля 2011 года.
- ↑ Medscape: Medscape Access. Emedicine.medscape.com. Дата обращения: 26 января 2013. Архивировано 18 января 2013 года.
- ↑ Литвицкий П. Ф. Моногенные формы патологии, наиболее часто встречающиеся в клинической практике — Фенилкетонурия // Клиническая патофизиология: учебник. — М.: Практическая медицина, 2015. — С. 92. — 776 с. — ISBN 978-5-98811-349-2.
- ↑ Умственная отсталость // Психиатрия: национальное руководство / под ред. Т. Б. Дмитриевой, В. Н. Краснова, Н. Г. Незнанова, В. Я. Семке, А. С. Тиганова. — М.: ГЭОТАР-Медиа, 2011. — С. 666. — 1000 с. — ISBN 978-5-9704-2030-0.
- ↑ 1 2 3 4 О. В. Кербиков, М. В. Коркина, Р. А. Наджаров, А. В. Снежневский. Разновидности олигофрений. Фенилпировиноградная олигофрения // Психиатрия. — 2. — М.: Медицина, 1968. — С. 403.
- ↑ Постановление Правительства РФ № 1719. static.government.ru (22 октября 2020). Дата обращения: 30 октября 2020. Архивировано 1 ноября 2020 года.
- ↑ LLLI: Кормление ребёнка, больного фенилкетонурией Архивная копия от 22 октября 2014 на Wayback Machine / Терри Гроссмайер, Breastfeeding a Baby with PKU Архивная копия от 25 августа 2016 на Wayback Machine / New Beginnings, сентябрь-октябрь 1998 года, 153—156 стр. (рус.)
- ↑ (недоступная ссылка с 22-08-2016 [3017 дней])National PKU News: Tips for Breastfeeding a Baby with PKU Архивная копия от 4 декабря 2008 на Wayback Machine (англ.)
- ↑ Принципы лечения детей, больных фенилкетонурией. — Екатеринбург, 2005, с. 14
- ↑ Opladen T, López-Laso E, Cortès-Saladelafont E, Pearson TS, Sivri HS, Yildiz Y, Assmann B, Kurian MA, Leuzzi V, Heales S, Pope S, Porta F, García-Cazorla A, Honzík T, Pons R, Regal L, Goez H, Artuch R, Hoffmann GF, Horvath G, Thöny B, Scholl-Bürgi S, Burlina A, Verbeek MM, Mastrangelo M, Friedman J, Wassenberg T, Jeltsch K, Kulhánek J, Kuseyri Hübschmann O (May 2020). "Consensus guideline for the diagnosis and treatment of tetrahydrobiopterin (BH4) deficiencies". Orphanet Journal of Rare Diseases. 15 (1): 126. doi:10.1186/s13023-020-01379-8. PMC 7251883. PMID 32456656.
{{cite journal}}: Википедия:Обслуживание CS1 (не помеченный открытым DOI) (ссылка)
| В библиографических каталогах |
|---|