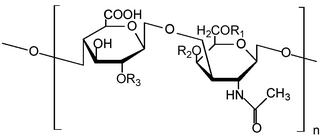
Гликозаминогликаны, мукополисахариды — углеводная часть протеогликанов, полисахариды, в состав которых входят аминосахара-гексозамины. В организме гликозаминогликаны ковалентно связаны с белковой частью протеогликанов и в свободном виде не встречаются.
Боле́знь Ха́нтера — одна из форм мукополисахаридоза, мукополисахаридоз 2-го типа, редкое рецессивное Х‑сцепленное генетическое заболевание из группы лизосомных болезней накопления. Возникает в результате дефицита ряда ферментов, что приводит к накоплению белково‑углеводных комплексов и жиров в клетках.

Мукополисахаридо́зы сокращённо МПС, или англ. MPS — группа метаболических заболеваний соединительной ткани, связанных с нарушением обмена кислых гликозаминогликанов, связанных недостаточностью лизосомных ферментов обмена гликозаминогликанов. Заболевания вызваны наследственными аномалиями обмена, проявляются в виде лизосомной болезни накопления: различных дефектов костной, хрящевой, соединительной тканей.
7-я хромосо́ма челове́ка — одна из 23 пар человеческих хромосом. Хромосома содержит более 158 млн пар оснований, что составляет от 5 до 5,5 % всего материала ДНК человеческой клетки. В настоящее время считается, что на 7-й хромосоме находятся от 1000 до 1400 генов.
3-я хромосо́ма челове́ка — одна из 24 человеческих хромосом, одна из 22 аутосом человека. Хромосома содержит почти 200 млн пар оснований, что составляет примерно 6,5 % всего материала ДНК человеческой клетки. В настоящее время считается, что на 3-й хромосоме находятся 1960 генов.

Стероидная сульфатаза (стероидсульфатаза) — сульфатаза человека, участвующая в метаболизме стероидов. Кодируется геном STS на X-хромосоме, расположенным вблизи псевдоаутосомной области.

Мукополисахаридоз IV типа или синдром Моркио — наследственное заболевание из группы лизосомных болезней накопления, впервые описано L. Morquio в 1929 году. Клинические проявления характеризуются значительными деформациями скелета, особенно грудной клетки. В отличие от других типов мукополисахаридозов, IV тип характеризуется отсутствием снижения интеллекта, помутнения роговицы, гепатоспленомегалии и гротескных черт лица.
Мезенхима́льные дистрофи́и — нарушения метаболизма, развивающиеся в строме органов

Гипертелоризм — ненормальное (увеличенное) расстояние между двумя парными органами. Обычно имеется в виду глазной гипертелоризм, для которого характерно увеличенное расстояние между внутренними углами глаз и зрачками. Это состояние необходимо отличать от телекантуса, при котором увеличено расстояние только между внутренними углами глазных щелей. Также характерными для этого состояния являются: увеличенная клиновидная кость, другие врождённые дефекты, возможно отставание умственного развития.

Лизосо́мные боле́зни накопле́ния — общее название группы весьма редких наследственных заболеваний, вызванных нарушением функции внутриклеточных органелл лизосом. Эти одномембранные органоиды являются частью эндомембранной системы клетки и специализируются на внутриклеточном расщеплении веществ: гликогена, гликозаминогликанов, гликопротеинов и других. Лизосомные болезни накопления вызываются генетически обусловленным дефицитом ферментов лизосом, что приводит к накоплению макромолекул, являющихся субстратом этих ферментов, в различных органах и тканях организма.

Синдро́м Санфили́ппо — группа клинически сходных редких наследственных заболеваний, относящихся к лизосомным болезням накопления. Заболевания связаны с дефицитом того или иного фермента, в результате чего в лизосомах накапливается один тип гликозаминогликанов — гепарансульфат. Нарушения, в соответствии с дефектом гена, разделяют на четыре формы. Синдром описан в 1963 году американским учёным-педиатром Сильве́стром Санфили́ппо с соавторами. Пациенты с синдромом Санфилиппо обычно доживают лишь до юности или 1-го периода зрелости.

Гаргоили́зм — характерная для болезни Пфаундлера — Гурлер и некоторых других типов мукополисахаридоза внешность: грубые черты лица: широкая и приплюснутая переносица, широко расставленные глаза, широкий с большими ноздрями нос, толстые губы и язык, открытый рот с маленькими редкими зубами; форма черепа нередко напоминает киль лодки. Заболевание обусловлено наследственной патологией соединительной ткани, проявляющейся поражением костей, суставов, глаз, внутренних органов и нервной системы. Понятие гаргоилизм берёт начало от фр. gargouille (гаргу́лья), обозначающего раструбы старинных водосточных труб средневековых соборов, украшавшиеся рельефными изображениями фантастических фигур с отталкивающим, причудливым лицом (химер).
Синдро́м Гу́рлер — тяжёлое наследственное заболевание из группы мукополисахаридозов, относящихся к лизосомным болезням накопления. Характеризуется недостаточностью альфа-L-идуронидазы — фермента лизосом, участвующего в катаболизме кислых мукополисахаридов, которые составляют основу межклеточного вещества соединительной ткани.
Синдро́м Шейе — тяжёлое наследственное заболевание из группы мукополисахаридозов, относящихся к лизосомным болезням накопления. Характеризуется недостаточностью альфа-L-идуронидазы — фермента лизосом, участвующего в катаболизме кислых мукополисахаридов, которые составляют основу межклеточного вещества соединительной ткани. Заболевание редкое, проявляется в детском возрасте.
Синдро́м Гу́рлер — Шейе́ — наследственное заболевание из группы мукополисахаридозов, относящихся к лизосомным болезням накопления. Характеризуется недостаточностью альфа-L-идуронидазы — фермента лизосом, участвующего в катаболизме кислых мукополисахаридов, которые составляют основу межклеточного вещества соединительной ткани. Заболевание редкое, проявляется в детском возрасте.

Мукополисахаридо́з I — группа метаболических заболеваний соединительной ткани, связанных с нарушением обмена кислых гликозаминогликанов, вызванных недостаточностью лизосомного фермента обмена гликозаминогликанов альфа-L-идуронидазы. Данная группа мукополисахаридозов связана с наследственной аномалией, вызванной генетически детерминированным дефектом гена, расположенного в локусе 4q16.3, которая наследуется по аутосомно-рецессивному типу. Проявляется в виде лизосомной болезни накопления, которая приводит к различным дефектам нервной, костной, хрящевой и соединительной тканей.
Синдром Марото — Лами — редкая наследственная болезнь, одна из форм мукополисахаридоза из группы лизосомных болезней накопления, биохимически связанная с дефицитом фермента лизосом N-Ацетилгексозамин-4-сульфатсульфатазы.
GM1-ганглиозидо́зы — редкие наследственные заболевания из группы лизосомных болезней накопления. Развитие клинической картины обусловлено дефектом или недостатком β-галактозидазы, который ведёт к нарушению метаболизма и накоплению субстратов (ганглиозида GM1, гликопротеинов и кератансульфата) главным образом в нервных клетках центральной и периферической нервной системы.
Синдро́м Сла́я — редкое наследственное заболевание из группы мукополисахаридозов, относящееся к лизосомным болезням накопления, характеризуется дефицитом фермента лизосом β-глюкуронидазы. В свою очередь, дефицит β-глюкуронидазы ведёт к накоплению определённых сложных углеводов (мукополисахаридов) во многих тканях и органах тела человека.
Мно́жественная сульфата́зная недоста́точность — очень редкое аутосомно-рецессивное наследственное заболевание из группы лизосомных болезней накопления. Болезнь развивается в результате недостаточности формилглицинсинтезирующего фермента, активирующего некоторые сульфатазы, среди которых три изофермента арилсульфатазы A, B, C и другие сульфатазы. Клиническая картина заболевания напоминает позднюю инфантильную форму метахроматической лейкодистрофии и мукополисахаридоза.