Альбинизм — наследственное заболевание, полное или почти полное отсутствие пигмента меланина или хлорофилла. Проявляется отсутствием нормальной вида окраски кожи, волос, шерсти, радужной и пигментной оболочек глаз, зелёных частей растений.

Кардиомиопатии — гетерогенная группа заболеваний миокарда, связанных с механической или электрической дисфункцией, которая обычно проявляется неадекватной гипертрофией или дилатацией. Кардиомиопатии могут как изолированно поражать только сердце, так и быть частью генерализованного системного заболевания, часто приводят к сердечно-сосудистой смерти или к инвалидизации, обусловленной прогрессирующей сердечной недостаточностью[].
Онкоге́н — ген, продукт которого может стимулировать образование злокачественной опухоли. Мутации, вызывающие активацию онкогенов, повышают шанс того, что клетка превратится в раковую клетку. Считается, что гены-супрессоры опухолей (ГСО) предохраняют клетки от ракового перерождения, и, таким образом, рак возникает либо в случае нарушения работы генов-супрессоров опухолей, либо при появлении онкогенов.

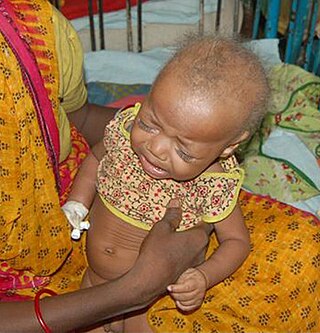
Болезнь Ниманна — Пика — это наследственное заболевание, вызванное нарушением липидного метаболизма и накоплением липидов, в частности сфингомиелина, в лизосомах клеток печени, селезёнки, лёгких, костного мозга и головного мозга. Заболевание относится к лизосомным болезням накопления и характеризуется аутосомально-рецессивным наследованием. Различают три типа заболевания: типы A, B и C.

Церулоплазмин — медь-содержащий белок (металлопротеин), присутствующий в плазме крови. В церулоплазмине содержится около 95 % общего количества меди сыворотки крови человека. Врожденный дефицит церулоплазмина приводит к дефектам развития головного мозга и печени. Был описан в 1948 году.
Меланоцитстимулирующие гормоны — гормоны средней, или промежуточной, доли гипофиза позвоночных животных и человека. По химической природе — полипептиды.
Болезнь Вильсона — Коновалова — врождённое или приобретенное нарушение метаболизма меди, приводящее к тяжелейшим поражениям центральной нервной системы и внутренних органов. Диагностируется у 5-10 % больных циррозом печени дошкольного и школьного возраста. Заболевание передаётся по аутосомно-рецессивному типу. Ген ATP7B, мутации которого вызывают заболевание, расположен на 13-й хромосоме.
3-я хромосо́ма челове́ка — одна из 24 человеческих хромосом, одна из 22 аутосом человека. Хромосома содержит почти 200 млн пар оснований, что составляет примерно 6,5 % всего материала ДНК человеческой клетки. В настоящее время считается, что на 3-й хромосоме находятся 1960 генов.
6-я хромосо́ма челове́ка — одна из 24 человеческих хромосом. Хромосома содержит более 171 млн пар оснований, что составляет от 5,5 до 6 % всего материала ДНК человеческой клетки. В настоящее время считается, что на 6-й хромосоме находятся от 1100 до 1600 генов.
Ацерулоплазминемия — редкое заболевание, вызванное мутациями гена, кодирующего синтез церулоплазмина. Данный белок, основная функция которого заключается в транспорте меди, также участвует в метаболизме железа, и мутации, нарушающие эту функцию церулоплазмина, приводят к токсичным отложениям железа в мозге, печени, поджелудочной железе.
Болезнь Менкеса — нарушение клеточного транспорта меди, при котором наблюдается замедление роста, патологии нервной системы, а также характерное закручивание волос[каких?], за что это состояние также называют «болезнью курчавых волос». Комплекс симптомов вызывается мутациями в гене, кодирующем АТФ-азу АТФ-7α, участвующую в поглощении меди из пищи и передаче ионов этого металла в другие клетки в трансмембранном переносе ионов меди.
Гефестин — белок человека, кодируемый геном HEPH на X-хромосоме. Назван в честь Гефеста, древнегреческого бога огня, покровителя кузнечного дела. Структура гефестина смоделирована по третичной структуре церулоплазмина, содержит гомологичный церулоплазмину активный центр и участки связывания меди. В отличие от церулоплазмина, в молекуле гефестина один из доменов является трансмембранным.

17-я хромосо́ма челове́ка является частью генома человека и насчитывает более 81 млн пар оснований, что составляет от 2,5 до 3 % всего ДНК-материала клетки. По разным оценкам, содержит от 1200 до 1500 генов.

20-я хромосо́ма челове́ка является частью генома человека и насчитывает более 63 млн пар оснований, что составляет от 2 до 2,5 % всего ДНК-материала клетки.
Рефлекс Штрюмпеля — патологический рефлекс проявляющийся в разгибании I пальца стопы при противодействии врача сознательной попытке больного согнуть ногу в коленном и тазобедренном суставах. Назван в честь немецкого невролога Адольфа фон Штрюмпеля, описавшего данный рефлекс в 1899 году.
Нонсенс-мутация — точечная мутация в последовательности ДНК, которая приводит к появлению стоп-кодона, в результате чего происходит преждевременная терминация синтеза нужного белка. Обычно такой фрагмент не может выполнять функции изначально синтезируемого белка.

JAG1 (CD339) — мембранный белок, лиганд для рецептора Notch1, играет роль на ранних и поздних этапах гемопоэза. Мутации белка вызывают синдром Алажилля 1-го типа.

GM2-ганглиозидо́з — тяжёлое наследственное заболевание, развивающееся в результате дефицита или недостаточной активности фермента гексозаминидазы и накопления в клетках ганглиозидов. Относится к лизосомным болезням накопления и имеет три варианта, связанные с мутациями в разных генах, которые оказывают вляние на активность общей гексозаминидазы. Два варианта заболевания больше известны под индивидуальными именами, полученными в честь авторов, впервые описавших их клиническую картину: болезнь Тея — Сакса, болезнь Сандхоффа. Третий вариант этой болезни носит название GM2 ганглиозидоз, вариант АБ. Болезнь Тея — Сакса вызвана мутацией в гене HEXA, кодирующий альфа-субъединицу гексоаминидазы А. Болезнь Сандхоффа вызвана мутацией в гене HEXB, кодирующий бета-субъединицу гексоаминидаз А и Б. GM2 ганглиозидоз, АБ вариант связан с нарушением в гене GM2, кодирующий белок-активатор GM2A.
Вителиформная макулярная дистрофия или вителиформная дистрофия — генетическое расстройство глаз, порождающее прогрессирующую потерю зрения, влияющее на макулярную область сетчатки, которая обеспечивает резкое центральное зрение, необходимое для получения мелких деталей изображения для чтения, вождения автомобиля, и распознавания лиц.
Болезнь Штаргардта или жёлтопятнистая абиотрофия сетчатки , является унаследованной формой ювенильной макулярной дегенерации, которая вызывает прогрессирующее ухудшение зрения, как правило, до точки официальной слепоты. Прогрессирование обычно начинается в возрасте от шести до двенадцати лет, и выравнивается вскоре после быстрого снижения остроты зрения. Несколько генов ассоциированы с этим расстройством. Симптомы обычно развиваются до возраста двадцати лет, и включают волнистое видение, слепые пятна, размытость, нарушение цветового зрения, и трудности с адаптацией при тусклом освещении.